All published articles of this journal are available on ScienceDirect.

Advanced Choroideremia Linked to a Rare Missense Variant in the CHM Gene
Abstract
Background
Choroideremia is an X-linked inherited chorioretinal dystrophy leading to blindness by late adulthood. Choroideremia is caused by mutations in the CHM gene, which encodes Rab escort protein 1 (REP1), a ubiquitously expressed protein involved in intracellular trafficking and prenylation activity.
Case Report
We report a 48-year-old male with an 18-year history of progressive night vision impairment. Fundoscopic examination revealed sharply demarcated regions of chorioretinal atrophy, primarily around the optic nerve. Bone spicules were observed throughout the retina, particularly at the margins of the atrophic areas, with preservation of the macula. Electroretinography (ERG) demonstrated a diminished response. Genetic testing confirmed a hemizygous missense variant in the CHM gene (CHM: NM_000390:exon11:c.1413G>C:p.Q471H), establishing the diagnosis.
Conclusion
This case, supported by clinical findings, multimodal retinal imaging, and genetic analysis, confirms that this CHM variant is pathogenic and contributes to choroideremia.
1. INTRODUCTION
Choroideremia (CHM, OMIM 303100) is an inherited chorioretinal dystrophy that manifests as a progressive degenerative disorder of the photoreceptor layer, retinal pigment epithelium (RPE), and choroid [1]. Current estimates place the prevalence of CHM between 1 in 50,000-100,000 people with a preponderance in the Finish population [1-5]. Choroideremia is an X-linked recessive condition that manifests in males, mostly with the symptoms of early night blindness followed by subsequent loss of peripheral vision, culminating in legal blindness in adulthood. CHM gene is located on chromosome Xq21.2, spans a DNA genomic sequence of 186,382 base pairs, consists of 15 exons, and is ubiquitously expressed [2]. The CHM gene encodes the Rab escort protein 1 (REP1), which plays a critical role in intracellular trafficking processes, such as protein transport and waste removal of the RPE. In choroideremia, the disease is restricted to the retina, likely due to the absence of REP2 in the retina, which compensates for REP1 deficiencies in other tissues [4]. Early stages of the disease are marked by peripheral pigmentary retinopathy, which progresses to chorioretinal atrophy with visible large choroidal vessels and scleral exposure [1]. Optical coherence tomography (OCT) often reveals increased central retinal thickness until the fourth decade, followed by a gradual reduction in most patients [6]. Full-field electroretinogram (ERG) demonstrates an early reduction in rod-driven responses and a more gradual but progressive cone dysfunction. ERG may be extinguished in advanced disease [1].
Despite advances in genetic diagnostics, choroideremia exhibits significant genetic heterogeneity, with mutations, including nonsense, frameshift, and splicing variants commonly reported in the CHM gene. However, the pathogenicity of some missense variants remains uncertain, warranting further investigation. This report presents a rare, missense variant in the CHM gene associated with advanced choroideremia, underscoring the clinical and molecular complexity of the disease.
2. CASE REPORT
A 48-year-old male presented with an 18-year history of progressive night vision loss in both eyes. His family history revealed consanguinity, though his parents and 11 siblings (five males and six females) displayed no ophthalmic symptoms. The symptoms experienced by the patient included minor deficiencies in his visual field, blurred vision, and poor night vision.
Upon examination, his best-corrected visual acuity (BCVA) was 20/40 in both eyes with -0.50 diopters. The anterior segment examination was normal, and the dilated fundus examination showed peripapillary chorioretinal atrophy extending to the equator, arteriolar attenuation, and bone spicules (Fig. 1).
Spectral-domain OCT scans (OCT, Heidelberg Engineering, Inc., Heidelberg, Germany) revealed mild disruption of the ellipsoid zone at the fovea, minor cystic changes, and choriocapillaris loss (Fig. 1). Visual field testing demonstrated severe constriction (Fig. 2), while the electroretinogram was nonrecordable. Fundus autofluorescence indicated limited RPE preservation and a speckled pattern corresponding to pigment clumps in the color image (Fig. 1). These clinical findings were compatible with choroideremia (6).
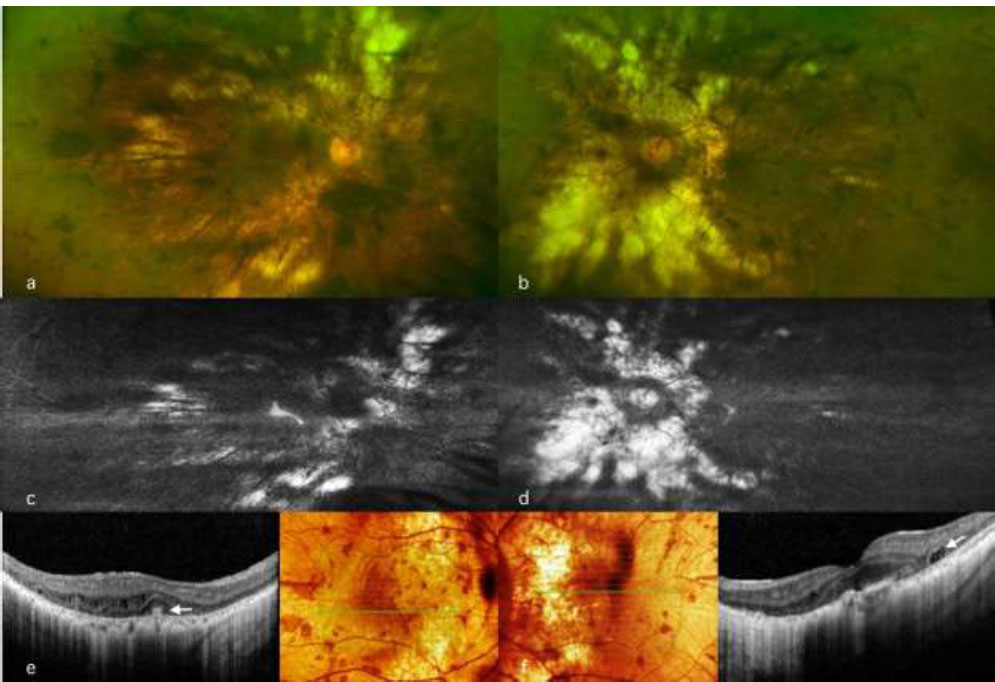
Multimodal retinal imaging in a 48-year-old male patient with the mutation in the (c.1413G>C:p.Q471H) CHM gene. Top left: (a) Wide-field fundus image of the right eye showing peripapillary chorioretinal atrophy extending to the equator with arteriolar attenuation and bone spicules. Top right: (b) Wide-field fundus image of the left eye showing similar findings to the other eye with more peripapillary chorioretinal atrophy. Middle: (c, d) Fundus autofluorescence of the right and left eyes highlighting the area of chorioretinal atrophy and a speckled pattern corresponding to areas, where there was visible pigment clumping in the color image. Bottom: (e, f) Trans-foveal optical coherence tomography (OCT) scan of both eyes showing mild disruption of the ellipsoid zone at the foveal area with minor cystic alterations (white arrow).
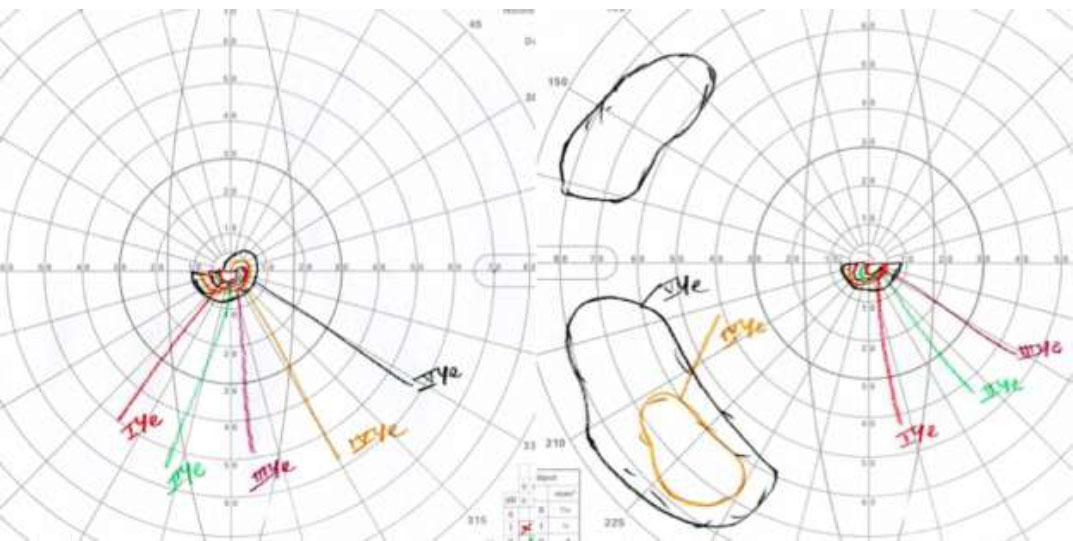
Goldmann visual fields of both eyes showing severely constricted fields in a 48-year-old male patient with the mutation in the (c.1413G>C:p.Q471H) CHM gene.
Genetic analysis using next-generation sequencing (NGS) and coding sequence identified a hemizygous missense variant in the CHM gene (CHM: NM_000390: exon11:c.1413G> C:p.Q471H), confirming the diagnosis. The genes included in this panel are available at https://genomebiology.biomedcentral.com/articles/10.1186/s13059-015-0693-2#Sec19. To the best of our knowledge, this specific CHM mutation and its clinical findings have not been reported in the literature.
3. DISCUSSION
Choroideremia (CHM) is a rare inherited retinal dystrophy caused by mutations in the CHM gene, which encodes Rab escort protein 1 (REP1) [4]. The rarity of choroideremia and the heterogeneity of CHM mutations make establishing genotype-phenotype correlations challenging. In this case, the identified hemizygous missense variant (CHM:NM_000390:exon11:c.1413G>C:p.Q471H) has not been previously reported in association with choroideremia. This mutation is not mentioned in GnomAD V2.1.1 (https://gnomad.broadinstitute.org, access date March 23rd, 2020). It is reported only once in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/, access date March 23rd, 2020) without any further information or any clinical correlation. However, it has not been reported before to be associated with CHM. This variant is classified as a Variant of Uncertain Significance (VUS), but several factors support its likely pathogenicity. The substitution at position 471 replaces glutamine with histidine, potentially altering the structure or function of REP1. While the residue at position 471 is not conserved across species, a nonsense mutation affecting the same amino acid (c.1411C>T, p.Q471*) has been classified as likely pathogenic in ClinVar. This suggests that changes in this position may have functional consequences [7].
Moreover, other missense mutations in the CHM gene have been linked to choroideremia, such as c.1679T>C (p.L550P) and c.1520A>G (p.H507R) [8]. These mutations disrupt REP1 function, leading to similar clinical phenotypes. Structural modeling of REP1 has demonstrated that missense variants can disrupt its interaction with Rab proteins or destabilize the protein, impairing its ability to facilitate intracellular trafficking [9]. While functional studies on the c.1413G>C variant are lacking, the severe clinical findings in this patient, including extensive peripheral chorioretinal atrophy, non-recordable electroretinogram, and severely constricted visual fields, align with the classic presentation of choroideremia, supporting the variant's pathogenicity [5, 9].
The clinical presentation of this patient further highlights the variability in disease progression among individuals with choroideremia. Despite an 18-year history of night vision loss, the patient retained relatively good central visual acuity (20/40 in both eyes) and exhibited only mild disruption of the ellipsoid zone at the fovea on spectral domain optical coherence tomography (SD-OCT). These findings are consistent with the natural history of the disease, in which central retinal function is preserved until the later stages, even as peripheral vision deteriorates significantly [1]. Choroideremia has previously been described to present in various ways, such as being similar to gyrate atrophy, retinitis pigmentosa, thioridazine hydrochloride retinal toxicity, Bietti’s crystalline dystrophy, and Kearns–Sayre syndrome [10]. Research conducted by Shiqiang et al. noted that 3.8% (6 out of 157) of cases diagnosed as RP families were later found to be misdiagnosed following genetic testing, where they were noted to have pathogenic mutations in the CHM gene [11].
The absence of a family history of choroideremia in this patient is noteworthy, given the X-linked inheritance pattern of the disease. This could be explained by incomplete penetrance or variable expressivity among female carriers or by the possibility of a de novo mutation in the CHM gene [4]. Segregation analysis could not be performed in this case due to the lack of genetic testing in other family members, which limits our ability to establish a definitive link between the c.1413G>C variant and the observed phenotype. However, the patient's consanguineous background may have contributed to the expression of this rare variant [10]. From a therapeutic perspective, the identification of a novel CHM variant has implications for gene therapy and other emerging treatments for choroideremia [8]. While no effective therapy exists for CHM, phase 3 clinical trials are currently taking place on gene therapy, and numerous pharmacological products and non-viral delivery methods are currently under development [11].
A limitation of our case report is the inability to perform segregation analysis on family members due to the unavailability of genetic testing. While segregation data would strengthen the evidence for pathogenicity, our conclusion is supported by clinical correlation, genetic analysis, and literature evidence. Future studies with family testing and functional validation may further clarify the significance of this variant.
In conclusion, this case contributes to the growing body of evidence on the genetic and clinical spectrum of choroideremia. It underscores the importance of integrating clinical findings, genetic analysis, and functional studies to characterize rare variants in the CHM gene. Further research is necessary to elucidate the mechanisms by which the c.1413G>C variant impacts REP1 function and to expand our understanding of genotype-phenotype relationships in this condition.
CONCLUSION
This case identifies a rare CHM gene variant (c.1413G>C, p.Q471H) associated with advanced choroideremia. Clinical findings, alongside genetic analysis, suggest its likely pathogenicity, though functional studies are needed for confirmation. Understanding such variants broadens the knowledge of choroideremia’s genetic and clinical phenotypes spectrum and informs future therapeutic approaches, including gene therapy.
AUTHORS’ CONTRIBUTIONS
It is hereby acknowledged that all authors have accepted responsibility for the manuscript's content and consented to its submission. They have meticulously reviewed all results and unanimously approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| ERG | = Electroretinography |
| RPE | = Retinal Pigment Epithelium |
| OCT | = Optical Coherence Tomography |
| CHM | = Choroideremia |
| VUS | = Variant of Uncertain Significance |
| SD-OCT | = Spectral Domain Optical Coherence Tomography |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was approved by the Institutional Review Board of Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia (Reference Number: RD/26001/IRB/0417-20).
HUMAN AND ANIMAL RIGHTS
All procedures performed in studies involving human participants were in accordance with the ethical standards of institutional and/or research committee and with the 1975 Declaration of Helsinki, as revised in 2013.
CONSENT FOR PUBLICATION
Written informed consent was obtained from the patient prior to writing this report.
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of the article will be available from the corresponding author [A.M.A] upon reasonable request.
ACKNOWLEDGEMENTS
Declared none.

