All published articles of this journal are available on ScienceDirect.

Retinoblastoma Presenting in a Child with Hypomelanosis of Ito
Authors Info & Affiliations
Abstract
Purpose:
To describe a case of a child with a known history of pigmentary mosaicism suggestive of Hypomelanosis of Ito presenting with unilateral leukocoria, who was ultimately diagnosed with retinoblastoma.
Methods:
A report of a 16-month-old girl with pigmentary mosaicism and unilateral retinoblastoma.
Results:
A previously healthy 16-month-old girl with a diagnosis of a mosaic hypopigmentation at the age of 6 months based on a linear and whorled pattern of skin hypopigmentation along the lines of Blaschko, presented with unilateral strabismus, leukocoria, retinal detachment, and sub-retinal exudation. Hypomelanosis of Ito and other similar neurocutaneous syndromes are known to be associated with abnormal retinal pigmentation, vascular abnormalities, and retinal detachment. Examination included a fluorescein angiogram, ultrasonography, and an MRI of the brain and orbits that demonstrated features consistent with retinoblastoma.
Given these findings and a flat electroretinogram, the eye was enucleated with final pathologic confirmation of retinoblastoma.
Conclusions:
Previously unreported presentation of unilateral retinoblastoma in a child with pigmentary mosaicism.
CASE REPORT
A 16-month-old girl presented with the concern for a “glassy” right eye that had recently been noted to deviate outward. The child’s general health and behavior were otherwise normal, but had been evaluated by a pediatric dermatologist 10 months prior for presence of a whorl-like pattern of hypopigmentation on her back, arms, and legs (Fig. 1). The impression was pigmentary mosaicism or Hypomelanosis of Ito (HI) and she was referred to a pediatric geneticist and ophthalmologist. Both ophthalmic examination and chromosomal analyses of her blood were normal at that time. A skin biopsy was considered, but not performed, to evaluate for suspected mosaicism.
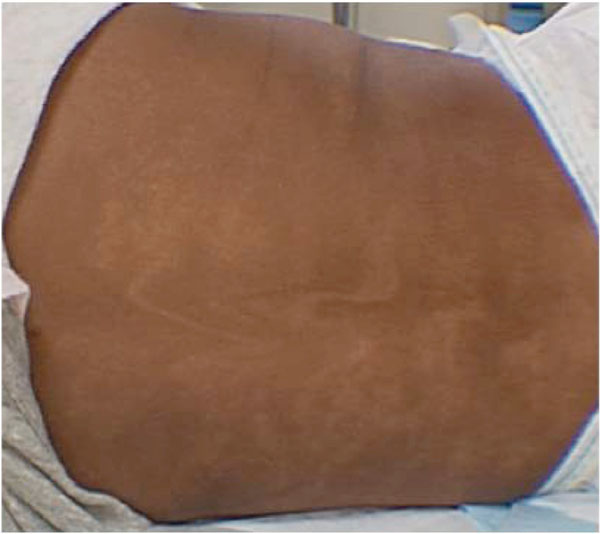
Color photograph of patient’s back demonstrating whorled pigmentation pattern following Blaschko’s lines.
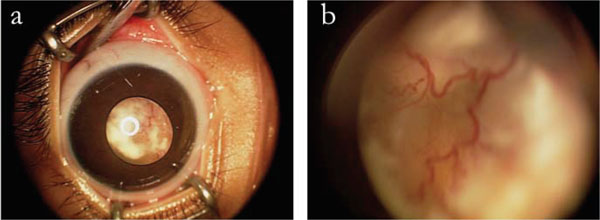
(a) External color photograph of anterior segment and creamy white lesion in posterior segment on right eye. (b) Detail of vascular lesion in posterior segment of right eye.
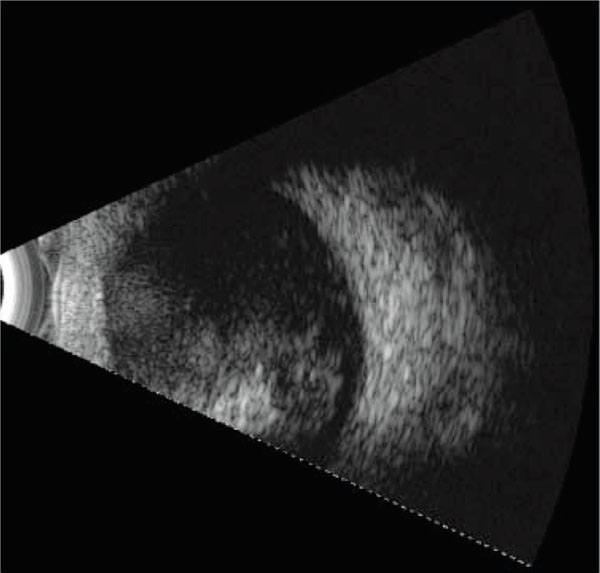
B-mode ultrasonography of the intra-ocular lesion of right eye demonstrating its large size and suggestion of calcification.
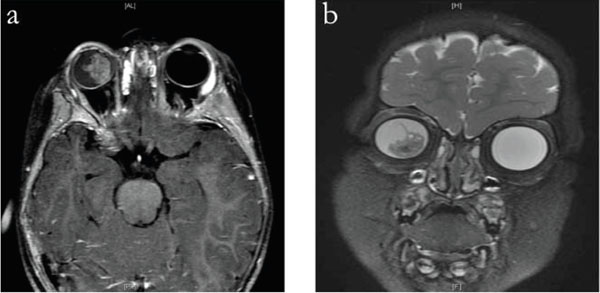
Axial T1 (a) and coronal T2 fat-saturated (b) MRI of brain and orbits demonstrating a large dense intra-ocular mass.

Photomicrograph of pathology specimen of enucleated right globe confirming the diagnosis of retinoblastoma (Hematoxylin-Eosin Stain at 200x).
On our examination (M.F.C.) of the right eye, she had no fixation, obvious leukocoria, +2 afferent pupillary defect in the right eye, and a 30-prism diopter exotropia. The right eye had a shallow anterior chamber without iris neovascularization, and a white retrolental mass with overlying retinal vessels (Fig. 2). The retrolental lesion appeared creamy white with multiple corkscrew vessels bowing inward, and was large enough to block any view to the posterior pole. B-scan ultrasonography demonstrated an intraocular mass occupying approximately 35% of the eye and a suggestion of a small amount of calcium within the mass. Examination of the left eye was normal.
Our differential diagnosis at this point was influenced by her prior dermatologic history as hypomelanosis of Ito has been reported to be associated with neurologic, ophthalmic, cardiac, renal, and craniofacial abnormalities [1, 2]. Despite this potential for an associated benign process, she was referred to an ophthalmic oncologist (D.A.) for our greater concern for retinoblastoma. This examination included fluorescein angiography that demonstrated abnormal retinal vasculature and areas of non-perfusion, B-scan ultrasonography that demonstrated a large highly reflective lesion with internal calcifications (Fig. 3), and an ERG that showed a nearly flat signal on the right and a normal signal on the left. An MRI of her orbits and brain showed a 1x1.4 cm enhancing mass in the right eye, with non-enhancing calcified foci overlying a retinal detachment with significant subretinal fluid (Fig. 4). There was no evidence of optic nerve or orbital pathology. Finally, she was referred for genetic testing and found not to carry the Rb1 mutation, thereby reducing but not eliminating the potential that she carries a germline mutation.
Our recommendation was to proceed with enucleation as the eye had no visual potential and a high risk of malignancy. The independent pathology report from the enucleated eye demonstrated retinoblastoma without scleral or optic nerve extension (R.F.) (Fig. 5). The child has since been followed without signs of recurrence on the affected side or lesion in the contralateral eye.
DISCUSSION
The term Hypomelanosis of Ito (HI), also known in the historical literature as incontinentia pigmenti achromians, has been used to describe an uncommon congenital neurocutaneous syndrome presenting in the first year of life. It is more common in girls, and is typified by a distinctive cutaneous whorled pattern of hypopigmentation, often on the trunk, in association with neurologic and/or developmental abnormalities. The hypopigmented streaks correlate with the lines of Blaschko, and suggest that a defect in neural crest cellular migration may account for the pattern. There are no preceding vesicles, pustules or verrucous plaques as are seen in infants with incontinentia pigmenti (IP.)
HI has been estimated to be present in between 1 in 3000 to 1 in 10,000 children in a general pediatric practice, but precise prevalence has been difficult to obtain because of its relative rarity [3-5]. Although the neurologic literature suggests a high association between mosaic hypopigmentation and neurologic abnormalities, the terms pigmentary mosaicism or hypopigmentation along the lines of Blaschko are preferred as patients may have cutaneous findings without associated neurodevelopmental abnormalities [6].
A wide range of chromosomal abnormalities in patients with pigmentary mosaicism have been reported, but an exact etiology is yet unclear. Approximately 50% of these children have an abnormal karyotype, and a mosaic pattern is often identified by skin biopsy with multiple X-chromosome lineages [7]. The majority of patients present without a family history of abnormal skin pigmentation, although there is a rare cohort with a known family history of skin lesions [7]. In patients with pigmentary mosaicim, chromosome abnormalities are best detected on skin fibroblast cultures, and may not be present in the blood. It remains a possibility that our patient harbors a deletion of the retinoblastoma gene in a mosaic pattern, which can best be demonstrated in fibroblast culture with FISH probes for the Rb1 gene or chromosomal microarrays.
HI has been associated with non-cutaneous findings including neurologic (both anatomic and functional), cardiac, genitourinary, musculoskeletal, and ophthalmic abnormalities. tourinary abnormalities include Ophthalmic abnormalities are reported in 20% of patients and involve the anterior and posterior segments, as well as nystagmus, refractive errors, and strabismus [8-10]. Anterior segment abnormalities include subtle iris hypopigmentation and corneal asymmetry to more profound changes such as an opaque cornea, cataracts, and Axenfeld-type anomalies. Posterior segment abnormalities include choroidal hypopigmentation and atrophy, optic atrophy, and retinal detachment.
The intraocular lesion in this case included several highly suspicious features for retinoblastoma including its creamy white color, extensive subretinal exudation and abnormal retinal vessels. However, we were also concerned that these lesions could be a variant of the pathology associated with incontinentia pigmenti (IP), known to be associated with retinal vessel abnormalities, retinal hypopigmentation, and rapidly progressive exudative retinal detachment: all seen on examination of our patient. Following extensive conversations with the child's mother, and despite the potential for an advanced but benign intraocular process, we recommended enucleation for a sightless eye and ultimately received pathologic confirmation of retinoblastoma This case illustrates a diagnostic dilemma in patients with neurocutaneous syndromes with known ophthalmic findings that could be a masquerade for retinoblastoma.
ACKNOWLEDGEMENT
None declared.
DISCLOSURES
The authors have no proprietary interest related to this work. Supported by NIH grant EY19474 (M.F.C.), by a Research to Prevent Blindness Career Development Award (M.F.C.), and The Fund for Ophthalmic Knowledge, Inc. (D.A.). M.F.C. is an unpaid member of the Scientific Advisory Board for Clarity Medical Systems (Pleasanton, CA).
CONFLICTS OF INTEREST
None declared.

