All published articles of this journal are available on ScienceDirect.

Diabetic Retinopathy and VEGF
Authors Info & Affiliations
Abstract
Diabetic retinopathy remains the leading vascular-associated cause of blindness throughout the world. Its treatment requires a multidisciplinary interventional approach at both systemic and local levels. Current management includes laser photocoagulation, intravitreal steroids, and anti-vascular endothelial growth factor (VEGF) treatment along with systemic blood sugar control. Anti-VEGF therapies, which are less destructive and safer than laser treatments, are being explored as primary therapy for the management of vision-threatening complications of diabetic retinopathy such as diabetic macular edema (DME). This review provides comprehensive information related to VEGF and describes its role in the pathogenesis of diabetic retinopathy, and in addition, examines the mechanisms of action for different antiangiogenic agents in relation to the management of this disease. Medline (Pubmed) searches were carried out with keywords “VEGF”, “diabetic retinopathy”, and “diabetes” without any year limitation to review relevant manuscripts used for this article.
INTRODUCTION
Diabetic retinopathy (DR), a well-known consequence of long-standing and poorly controlled diabetes mellitus (DM), causes significant vision loss and blindness in the human population. DR currently affects approximately 150 million people worldwide, and the World Health Organization projects that the number of people affected will double by the year 2025 [1].
In patients with DM, the prevalence of any form of diabetic retinopathy is approximately 24% [2]. Diabetic macular edema (DME) is the major vision-threatening complication of DR. The Wisconsin Epidemiologic Study has reported that the prevalence of DME in diabetics of 15-year duration is approximately 20% in patients with Type 1 DM and 25% in patients with Type II DM that are on treatment [3]. Patients without macular edema can still maintain good vision even if they have advanced stages of the disease. Therefore, management of DME has become crucial for ophthalmologists in preventing vision loss to their diabetic patients.
Macular edema is diagnosed clinically by documenting retinal thickening and/or hard exudates using biomicroscopic examination. The Early Treatment Diabetic Retinopathy Study (ETDRS) has described a specific type of DME called “Clinically Significant Macular Edema” (CSME) for which laser treatment is indicated [4]. Based on angiographic findings, diabetic maculopathy has been classified as either focal or diffuse, and ischemic or non-ischemic. Diabetic maculopathy caused by leaking microaneurysms is classified as focal, while leakage at the level of the capillary beds leads to diffuse. The visual prognosis for patients with the ischemic type is very poor as there is deprivation of blood to the retina in ischemic zones.
Diabetic retinopathy is a disease of the retina, but similar vascular and ischemic changes can be found in other parts of the eye in the more advanced stages. Profound ischemia of the retina leads to neovascularization (growth of new vessels) of the disc (NVD), the retina (NVE), the iris (NVI) and the angle (NVA). NVA can lead to neovascular glaucoma (secondary to closure of the angle). For many years these conditions have been treated by panretinal photocoagulation (PRP) of the retina, which causes regression of the abnormal new vessels on the iris and angle [5]. In advanced cases of neovascular glaucoma where there is synechial closure of the angle, a surgical shunt such as a Baerveldt tube shunt or Ahmed glaucoma valve must be placed to facilitate aqueous drainage. Currently, anti-VEGF drugs are being investigated for the treatment of rubeosis. The RAVE (Rubeosis Anti-VEGF) clinical trial for Ischemic Central Retinal Vein Occlusion (CRVO) utilizes monthly ranibizumab injections for nine months to determine if this treatment can prevent the development of neovascular glaucoma in eyes with CRVO.
TREATMENT OF DIABETIC RETINOPATHY
Intensive glycemic control is one of the two important ways to reduce the risk of vision loss from DME. The Diabetes Control and Complications Trial [6] and UK Prospective Diabetes Study [7] showed that intensive blood glucose control reduced the risk of development of DME by 23%.
Laser photocoagulation is the second most important course of action to prevent vision loss from DME. Clinical trials such as the Diabetic Retinopathy Study (DRS) and ETDRS have shown that timely laser intervention can reduce the loss of vision by 90% [8, 9]. For proliferative diabetic retinopathy, PRP laser is the treatment of choice as demonstrated in several clinical trials. Unfortunately, the areas of retina treated with PRP laser exhibit scarring and loss of function.
In eyes of patients with DME, corticosteroids inhibit the expression of VEGF and decrease angiogenesis [10]. Platelet-derived growth factor (PDGF) is a proinflammatory cytokine that induces the expression of the VEGF gene, an effect that is downregulated by corticosteroids [11]. Intravitreal injections of triamcinolone acetonide (IVTA) are now commonplace and widely used for treatment of retinal disorders. In spite of being a fairly safe procedure, intravitreal steroid injections carry an inherent risk of major complications such as cataract, glaucoma, endophthalmitis (both infective and sterile), vitreous hemorrhage, and retinal detachment.
Visual outcomes are generally poor in advanced cases of DR with complications such as long-standing vitreous hemorrhage (VH), tractional retinal detachments (TRD), and fibrovascular proliferation. In these instances, the surgical approach is the procedure of choice [12]. The Diabetic Retinopathy Vitrectomy Study (DRVS) was a National Eye Institute sponsored, multicenter clinical trial program that established treatment guidelines for patients with advanced proliferative diabetic retinopathy. After four years of follow-up, the conclusions were that an early vitrectomy provided a greater chance for prompt visual recovery in eyes with recent severe vitreous hemorrhage.
Despite the presence of varied therapeutic modalities, vision loss occurs at an alarming rate due to DME. The ETDRS showed that despite laser treatment, 12% of subjects continued to have moderate visual loss. In addition, laser treatment is a destructive procedure that does not take into account the pathophysiology of DR and can cause tissue damage itself.
Recently, interest has developed regarding use of anti-VEGF in cases of DME. Although adverse effects such as nonfatal cerebrovascular accidents and myocardial infarction have been described [13], anti-VEGF agents are proving to be far superior to the laser treatments which are associated with extensive resultant tissue damage. The incidence of complications such as glaucoma and cataract encountered with the intravitreal injections of corticosteroids can be avoided with the use of anti-VEGF. For these reasons, they are becoming the preferred choice of treatment in cases of DME with coexisting glaucoma and in cases recalcitrant to lasers and intravitreal steroids.
Diabetic patients with concomitant hypertension are at a higher risk of developing DME and DR. Moreover, the DR in these patients tends to progress much more rapidly [14, 15]. The renin-angiotensin-aldosterone system (RAAS), which regulates blood pressure and maintains water balance, has a direct connection to the pathologic microvascular changes occurring in DR [16]. Angiotensin II, the major bioactive product of the RAAS, acts as an endocrine, paracrine, and intracrine hormone, and is a powerful vasoconstrictor of arterioles. In vitro studies have shown that angiotensin II directly stimulates the secretion of VEGF in cultured smooth muscle cells [17] and in cardiac endothelial cells [18]. Therefore, constriction of the retinal microvasculature could result in local increased VEGF production within the retina, which in turn could lead to angiogenesis [17, 19]. The detailed pathway linking angiotensin II to increased synthesis of VEGF is yet to be elucidated, but hypoxia-inducible factor (HIF-1α) has been shown to be involved at the intracellular signaling pathway level.
HISTORY OF THE VEGF MOLECULE
In 1948, Isaac Michaelson proposed that in the development of pathological angiogenesis, a key event was the synthesis and release of a diffusible angiogenic factor called “Factor X” by the ischemic retina [20]. Later in 1971, Judah Folkman presented the idea of inhibiting angiogenesis as a means to treat cancer, which then led to the discovery of anti-angiogenic factors [21]. Nearly forty years passed before the originally proposed “Factor X” could be isolated and identified. In 1983, Senger, Dvorak and colleagues discovered a key mediator involved in angiogenesis to be a protein secreted from a guinea pig tumor cell line. It had a potent capacity to induce vascular leakage and was named vascular permeability factor (VPF) [22]. In 1989, Napoleone Ferrara and colleagues identified a molecule present in bovine pituitary follicular cells that promoted the proliferation of endothelial cells and called it vascular endothelial growth factor (VEGF) [23]. Subsequently, the cloning of VPF by Daniel Connelly et al. and VEGF by Ferrara et al. confirmed that the two factors were the same protein [24, 25]. In 1992, two independent studies demonstrated that hypoxia could upregulate VEGF expression [26, 27]. Evidence from clinical studies supported the critical role of VEGF in ocular neovascularization. It was demonstrated that vitreous samples of patients with active proliferative retinopathies associated with diabetes and venous occlusive disorders had elevated VEGF levels [28].
BACKGROUND ON THE VEGF MOLECULE
Vascular endothelial growth factors (VEGF) are a subfamily of growth factors that function as signaling proteins for both vasculogenesis (the de novo formation of the embryonic circulatory system) and angiogenesis (the growth of blood vessels from pre-existing vasculature). VEGF is secreted primarily from retinal pigmented epithelial cells, pericytes, astrocytes, müller cells, glial cells, and endothelial cells. VEGF has several members including VEGF-A, VEGF-B, VEGF-C, VEGF-D and placental growth factor (PGF). VEGF-A is a 36-46 kDa glycosylated protein derived by alternate splicing of mRNA from a single, 8-exon VEGF gene [29]. VEGF-A is expressed as five mRNA splice variants – isoforms 121, 145, 165, 189, and 206, depending upon the number of amino acids present. Larger isoforms (isoforms 189 and 206) have increased heparin binding ability, which reflects their ability to bind to cell surfaces and basement membranes. Unfortunately, the larger isoforms also have limited or no diffusibility. Smaller isoforms (isoform 121 and 145) are readily diffusible but have limited heparin binding ability. Isoform 165 has intermediate levels of diffusibility and heparin binding. VEGF 165 is the principle isoform secreted, and is critical for both development and pathological angiogenesis [30]. The other VEGF members probably have a greater role to play in tumors and cancers than in DR [31].
VEGF PRODUCTION
VEGF production is induced in response to ischemia or hypoxia (Fig. 1). Tissue hypoxia leads to the production of a DNA binding protein called hypoxia-inducible factor 1 (HIF-1). HIF-1 is a heterodimeric, basic helix-loop-helix protein that consists of two subunits: the constitutively-expressed HIF-1 beta subunit and the growth factor regulated HIF-1 alpha subunit [32]. In addition to playing a crucial role in VEGF production, HIF-1 is also responsible for the upregulation of the human erythropoietin gene in hypoxic cells, which is another adaptation to tissue hypoxia [33]. HIF-1 binds to the VEGF gene and initiates the transcription process. This leads to the generation and accumulation of VEGF mRNA by both increased mRNA transcription and decreased mRNA degradation [34], eventually leading to the intracellular accumulation of VEGF molecules. A chaperone protein called ORP 150 (oxygen regulated protein) is responsible for the transfer of VEGF from the endoplasmic reticulum to Golgi bodies. The expression of ORP150 mRNA is also induced by hypoxia [35].
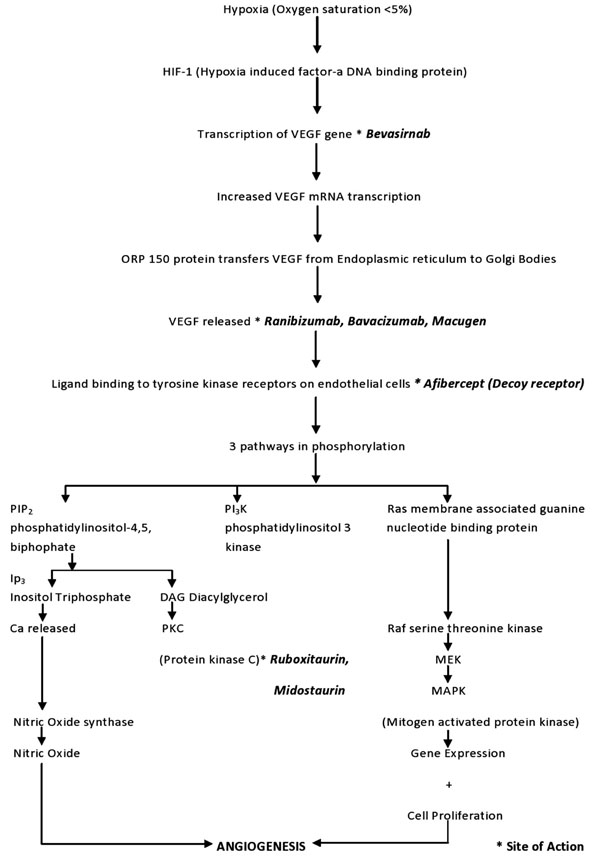
Flowchart of the VEGF cascade showing the molecular chain of events that occur after retinal tissue hypoxia culminating in angiogenesis.
VEGF ACTION
All members of the VEGF family stimulate cellular responses by binding to tyrosine kinase receptors on the endothelial cell surface, causing them to dimerize and become activated through transphosphorylation. VEGF-A has two types of receptors: VEGF receptor 1 (VEGFR-1 or Flt-1) and 2 (VEGFR-2 or KDR). In humans, VEGFR-1 is a protein that is encoded by the Flt-1 gene [35] and VEGFR-2 is a kinase insert domain receptor (KDR, a type III receptor tyrosine kinase) that is encoded by the KDR gene [36]. VEGFR-2 seems to be the primary receptor that mediates almost all of the cellular responses to VEGF-A. The VEGF receptors have three parts: an extracellular portion consisting of seven similar immunoglobulin-like domains, a single transmembrane hydrophobic spanning region, and an intracellular portion containing a split tyrosine kinase domain [37]. When the VEGF molecule binds to the extracellular portion of the VEGF receptors, the intracellular portion causes phosphorylation of the tyrosine amino acid residues and a cellular signal is transduced. This transduction in turn induces a cascade of intracellular signaling pathways. One of the pathways induced within the cells by VEGF is the protein kinase C (PKC) pathway [38]. The other pathways contribute to angiogenesis by synthesis of nitric oxide and induction of cellular proliferation.
VEGF INDUCED ANGIOGENESIS IN DR
VEGF is an important factor in the development of both proliferative DR and DME [39], altering retinal capillary permeability by increasing the phosphorylation of proteins involved with tight-junctions such as zonula occludens [40]. The VEGF induction activates mitogen-activated protein (MAP), resulting in endothelial cell proliferation. This cascade coincides with the activation of the phophatidylinositol 3-kinase (PI3)/Akt pathway after VEGFR-2 induction [33]. VEGF-A stimulates endothelial cells to release matrix metalloproteinases (MMPs) and urokinase-type plasminogen activator, resulting in the degradation of basement membranes and making cell migration possible. The activated endothelial cells express integrins such as αvβ3 and αvβ5, which aid in the migration through the degraded matrix [41]. Proliferation and migration of endothelial cells is followed by synthesis of basement membranes for the newly formed capillaries. The stability of these capillaries is achieved by recruitment of pericytes and smooth muscle cells that are regulated by platelet-derived growth factor (PDGF) [42].
VEGF AS A THERAPEUTIC TARGET
Much work has been done to study the mechanisms of VEGF in hopes of reducing neovascularization and vessel leakage by blocking its effects. Fig. (1) shows the sites of action of the drugs that target abnormal neovascularization.
Anti-VEGF Antibodies:
Commercially, there are two drugs available in this class. Ranibizumab (Lucentis, Genentech Inc., San Francisco, CA) is a humanized monoclonal antibody fragment (Fab, fragment antigen-binding) retrieved from the parent molecule of bevacizumab (Avastin, Genentech Inc., San Francisco, CA) against VEGF-A [43].
Ranibizumab was designed for intra-ocular use as a smaller antibody fragment for better intraocular penetration into the retina. It was approved in 2006 by the US Food and Drug Administration (USFDA) to be used for intravitreal injections in the treatment of neovascular (wet) age-related macular degeneration (AMD). The treatment protocol most commonly followed is three intravitreal injections given at intervals of four weeks. The retina is then evaluated and the need for further treatment is determined based upon the status of neovascularization. Pilot studies with intravitreal ranibizumab have also shown decrease in mean retinal thickness and improved vision in patients with DME [44]. In three well designed, phase II or III trials (RESOLVE [45], RIDE and RISE [13], READ 2 [46, 47]), 1–2 year treatments with ranibizumab were found to be more effective than sham or focal/grid laser therapy in improving best corrected visual acuity (BCVA) and reducing macular thickness in patients with visual impairment associated with DME. Along with these studies, two other well designed phase III trials (RESTORE [48] and DRCR.net [49-52]) showed that one year of treatment with ranibizumab as an adjunct to laser therapy was more effective than laser therapy alone in improving BCVA and macular thickness in patients with visual impairment associated with DME. Ranibizumab alone or as an adjunct to laser therapy showed improvement at the first follow-up visits in these studies, and were associated with gains in vision-related quality of life, assessed using the National Eye Institute Visual Functioning Questionnaire-25. The ocular and non-ocular adverse events were similar to the patients with wet age-related macular degeneration or retinal vein occlusion that had received ranibizumab.
Bevacizumab (Avastin, Genentech Inc., San Francisco, CA) is a full length humanized monoclonal antibody against all isoforms of VEGF-A and binds to the VEGF molecule to prevent receptor binding. It has the USFDA approval for intravenous treatment for metastatic colon cancer. Although bevacizumab does not have USFDA approval for intravitreal use for ocular diseases, it has been used off label and is very effective in inhibiting neovascularization associated with various retinal proliferative vascular diseases such as CNV, proliferative diabetic retinopathy, neovascular glaucoma, diabetic macular edema, and macular edema secondary to retinal vein occlusions. The DRCR phase II showed a more favorable outcome for bevacizumab treated patients at three weeks compared to laser photocoagulation alone [53]. The BOLT study also favored bevacizumab over laser photocoagulation at one year [54, 55].
Inhibitors of Intracellular Transcription:
Bevasiranib (OPKO, FL, USA) is the first small interfering RNA (siRNA) drug designed to silence the genes that produce VEGF, which is largely believed to be responsible for causing DME. The RNAi assessment of bevasiranib in DME or RACE trials was a pilot phase II investigation of the safety and preliminary efficacy of the drug in patients with DME. This 48 patient, multicenter, double-masked, randomized trial studied 3 dose levels of bevasiranib, and showed a trend of decrease in macular thickness between weeks 8 and 12. In addition, higher dosages resulted in a larger reduction in thickness than the lowest dose [56].
Inhibitors of Extracellular VEGF:
Pegaptanib sodium (Macugen, Eyetech Pharmaceuticals Inc., New York, NY and Pfizer Inc., New York, NY), a RNA aptamer, was one of the earliest anti-VEGF drugs which the USFDA approved for the treatment of neovascular AMD. It binds and blocks isoform 165 of VEGF [57]. The Macugen Diabetic Retinopathy Study Group conducted a phase II randomized controlled trial of Pegaptanib for fovea-involving DME. After 36 weeks follow-up, the Pegaptanib-treated eyes had better visual acuity, more reduction in central retinal thickness, and less need for laser photocoagulation compared to the sham group. More recently, a phase II/III randomized controlled trial reported that Pegaptanib therapy was associated with improved visual outcomes in patients with DME for up to 2 years [58, 59].
Inhibitors of VEGF Receptor Expression:
Aflibercept (VEGF-Trap, Regeneron Pharmaceuticals Inc., Tarrytown, NY and the Sanofi-aventis Inc., Paris, France) is a total human recombinant fusion protein that binds with all forms of VEGF-A and placental growth factor (PGF) [60]. The Regeneron and Bayer Inc. (Leverkusen, Germany) companies are developing the drug for ocular use. The DA VINCI study showed favorable outcomes when compared to laser photocoagulation in cases of DME. The outcome of the VEGF Trap-Eye trial at the primary endpoint of 24 weeks, as well as through the 52 week follow-up period, showed statistically significant improvement in visual acuity with the agent. Additionally, reduction in the optical coherence tomography (OCT) retinal thickness was noted. At the end of 6 months, patient eyes treated with aflibercept showed a gain of 8.5 to 11.4 letters on average, compared with 2.5 letters for the laser treated group. Moreover, at the end of 52 weeks, aflibercept treated eyes gained 9.7 to 13.1 letters as compared with a one-letter loss for laser treated eyes. At the end of one year, 45% - 71% of patient eyes gained 10 letters or more, and 24% - 46% of eyes gained 15 letters or more. The visual improvement was accompanied with reduction of OCT retinal thickness and was well tolerated by the patients. Presently, a phase III randomized clinical trial is currently underway to investigate the efficacy of VEGF Trap-Eye therapy for diabetic macular edema [61,62].
Inhibitors of Intracellular Signaling Cascade Activating VEGF:
Midostaurin (PKC412, Fermentek Biotechnology, Jerusalem, Israel) is a potent inhibitor of multiple isoforms of protein kinase C (PKC), VEGF-R2, PDGFR (platelet-derived growth factor receptor), KIT tyrosine kinase (a receptor that binds stem cell factor) and Flt-3 tyrosine kinases [63]. Midostaurin blocks the VEGF receptor signaling and thereby prevents retinal neovascularization [64]. PKC 412 has shown inhibition of ischemia-induced neovascularization in an animal model [65], however, early pharmacodynamic studies have shown some adverse outcomes, specifically revealing its lack of specificity. The principle toxicities of PKC412 are nausea/vomiting and fatigue. The best tolerated dose was found to be 150mg/d for chronic administration [66]. Even at this dose it was hepatotoxic, thus shifting focus to a more selective drug, ruboxistaurin [67].
Protein Kinase C (PKC) Beta Inhibitor:
Ruboxistaurin (Eli Lilly and Co., Delaware, Indianapolis) is an orally administered PKC beta isoform-selective inhibitor. It is an investigational drug for the treatment of moderate to severe non-proliferative DR. The PKC-Diabetic Retinopathy Study (PKC-DRS) was formulated to evaluate the effect of ruboxistaurin on visual loss in patients with moderately severe to very severe non-proliferative DR. It showed a reduction in the incidence of both moderate visual loss (doubling of the visual angle) and sustained moderate visual loss (for 6 months). The PKC-DME Study (PKC-DMES) also indicated reduced progression of DME with ruboxistaurin, although this was a secondary endpoint. Ruboxistaurin has not yet received approval from the USFDA [68-71].
SUMMARY AND CONCLUSION
Laser photocoagulation still remains the gold standard treatment for proliferative DR and DME, but it is not effective in many patients. Recently, retina specialists are inclined to use anti-VEGF agents for the management of DME. Although only some of these drugs are FDA approved for intraocular use (i.e., ranibizumab, pegaptanib), many are currently being used off label in clinical practices (i.e., bevacizumab). These anti-VEGF drugs are used as adjunct therapies to laser treatment, but there is an increasing trend towards using them as primary treatment for diabetic macular edema.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
This work was supported by Discovery Eye Foundation, Guenther Foundation, Iris and B. Gerald Cantor Foundation, Polly and Michael Smith Foundation, Lincy Foundation, Research To Prevent Blindness.

