All published articles of this journal are available on ScienceDirect.

X-linked Retinoschisis Associated with Retinitis Punctata Albescens Caused by a Mutation in the RS1 Gene: A Family Study
Abstract
Purpose:
To describe the clinical and genetic characteristics (mutation in RS1 gene) of a Spanish family with X-linked retinoschisis (XLRS) associated with retinitis punctata albescens (RPA).
Methods:
The detailed ophthalmological examination included best corrected visual acuity (BCVA), colour and autofluorescence photography, fluorescein angiography, optical coherence tomography and electrophysiology tests. A next-generation sequencing (NGS) strategy was applied to the index patient, and then sequenced in an Illumina NextSeq500 system. Candidate variants considered to be disease-causing in the patient were confirmed and segregated in the family by Sanger sequencing.
Results:
We have studied three siblings of 54, 59 and 50 years old. Two of them presented with macular foveoschisis and a whitish mottling of the pigment epithelium in the peripheral and equatorial retina, while the other had macular atrophy. Electroretinography revealed a reduced b-wave, while a-wave remained unchanged. Mutation in RS1 (c.98G>A; p.Trp33*) was identified as the cause of the disease.
Conclusion:
XLRS is a genetic disease that leads to irreversible visual loss. We describe an unusual phenotype manifestation of a known mutation.
1. INTRODUCTION
X-linked retinoschisis (XLRS) is a recessive, macular and peripheral retinal dystrophy that may be the result of any number of mutations in the retinoschisin 1 (RS1) gene. The RS1 gene encodes for the retinoschisin protein, which is involved in cell adhesion and neural conduction and is secreted primarily by photoreceptors and bipolar cells. This disease most commonly occurs in males. Although the pathophysiologic features of schisis cavity formation have not been fully elucidated, several mechanisms have been proposed, including vitreous traction on the susceptible retina with defective retinoschisin and a defect in the interaction between retinoschisin and intracellular Na+/K+ ATPase pumps leading to extracellular fluid accumulation in schisis cavities [1]. To date, according to the Human Gene Mutation Database (HGMD; 2018.2 version, https://portal.biobase-international. com), 251 different mutations in this gene are known to cause XLRS [2].
XLRS is characterized by foveal retinoschisis, which occurs in nearly 100% of patients, whereas peripheral schisis is present in 50% of patients with XLRS [2]. The most recent classification divides XLRS into 4 distinct clinical phenotypes: type 1, foveal; type 2, foveolamellar; type 3, complex; and type 4, foveal-peripheral [3]. Clinical diagnosis is not easily determined in certain cases because of the wide range of phenotypes that may include macular and retinal degeneration and secondary complications such as vitreous hemorrhage and retinal detachment; thus, genetic diagnosis is helpful. The differential diagnosis includes Goldmann-Favre syndrome, Eales disease, retinal periphlebitis, Wagner syndrome, senile retinoschisis, and familial exudative vitreoretinopathy [4].
2. METHODS
This study involved a Spanish family comprising 3 affected siblings. The study conformed to the tenets of the Declaration of Helsinki (Edinburgh, 2000) and was approved by the Institutional Review Boards of the University Hospitals Virgen del Rocio and Virgen Macarena, Seville. An informed consent form for clinical and molecular genetic studies was signed by all participants.
2.1. Ophthalmic Examinations
Ophthalmic examinations included Best-Corrected Visual Acuity (BVCA) by Snellen 20-feet test and refractive error test. After pupil dilation with 1% tropicamide, colour and autofluorescence fundus photographs were taken and fundus fluorescein angiography (FFA) was performed with retinal camera Topcon TRC-NW8 (Topcon Medical System, Oakland, NJ, USA). Using the DRI OCT Triton system (Topcon Medical System, Oakland, NJ, USA), a 6 × 6 mm three dimensional (3D) optical coherence tomography (OCT) macular cube scan and 12 radial B-scans were performed in both eyes of each patient. The average macular thickness and the average retinal nerve fiber layer thickness were measured.
2.2. Neurophysiology Tests
Electroretinography (ERG) and Visual Evoked Potential (VEP) were also obtained.
2.3. Genetic Tests
The genomic DNA of the 3 individuals in the family were isolated from peripheral blood using automated procedures (ChemagicTM 360, Waltham, MA, USA). The index patient underwent targeted next generation sequencing (NGS) that selectively captured 99 inherited retinal dystrophies (IRD) genes using SeqCap® EZ Choice Enrichment Kit (Roche NimbleGen, Madison, WI, USA), which were sequenced on the NextSeq 500 sequencer (Illumina, San Diego, CA, USA) with 150 bp paired-end reads. A validated custom bioinformatic pipeline was used for the alignment, variant calling, and variant annotation of the NGS data (Unpublished data). Copy Number Variations (CNVs) were identified using the coverage command of Browser Extensible Data (BED) tools. Candidate variants considered to be disease-causing in the patient were confirmed and segregated in the family by Sanger sequencing according to the 3730 DNA Analyzer manufacturer’s protocols (Applied Biosystems, Foster City, CA, USA). The nomenclature of variants was adjusted to the Human Genome Variation Society (http://varnomen.hgvs.org/) guidelines using Mutalyzer (https://mutalyzer.nl/).


3. RESULTS
3.1. Ophthalmic Examinations
The first of the siblings studied was a 54-year-old male with no family history. He had progressive loss of vision in both eyes since 2011. Vasculitis posterior uveitis with bilateral Cystic Macular Edema (CME) was suspected. He was treated with topical Nonsteroidal Anti-Inflammatory Drugs (NSAIDs), topical, oral, and intravitreal corticosteroids, and systemic immunosuppressants (cyclosporine, adalimumab, tocilizumab) without visual improvement or resolution of the CME.
At the initial consultation, the patient presented BVCA of 20/40 in the right eye and 20/32 in the left eye. In the anterior pole, no inflammatory signs were observed, while fundoscopy revealed bilateral whitish mottling of the pigment epithelium in the peripheral and equatorial retina in 360º as well as foveal images in “flower petals.”
The second sibling studied was a 59-year-old male without any previous ophthalmologic review who presented with long-term visual loss BVCA of 20/400 in both eyes. He presented with whitish mottling of the pigment epithelium in the peripheral and equatorial retina like his brother. The last of the siblings studied was a 50-year-old man with similar findings by fundoscopy of macular foveoschisis.
3.2. Imaging Techniques
The existence of cystic spaces in the internal nuclear layer, correspondent with macular foveoschisis, was confirmed by OCT and FA in the first and last of the siblings studied. FA revealed hyperfluorescent mottling scattered in the fundus with small hypofluorescent areas of pigmented deposits in the peripheral retina. This mottling was hyperautofluorescent by autofluorescence (Fig. 1). However, the second sibling did not present schisis in OCT but an area of central atrophy (Fig. 2).
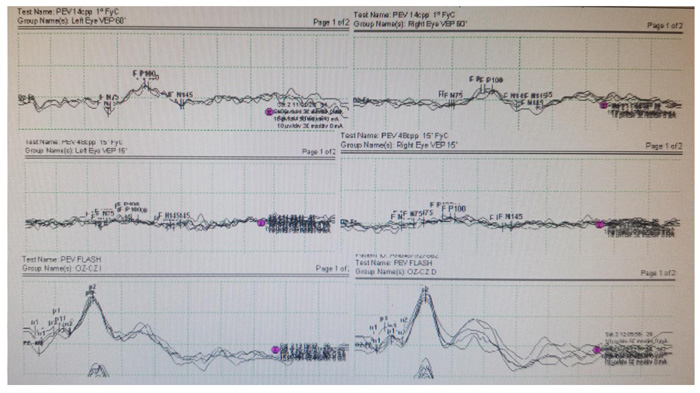
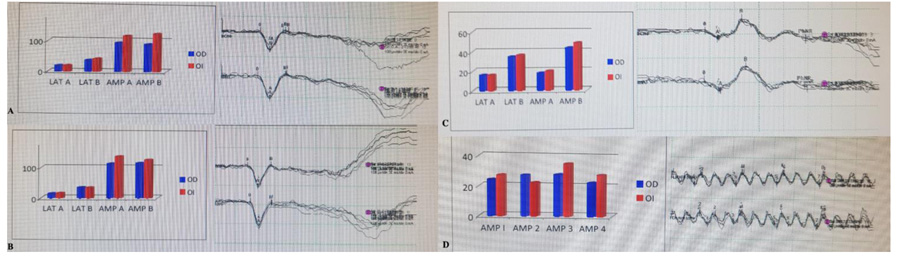
3.3. Neurophysiology
The electrophysiology tests were performed according to the recommendations of the International Society for Electrophysiology of Vision (ISCEV). VEP revealed latency of normal parameters for central vision fibers (Fig. 3), while ERG demonstrated depressed responses of cones, middle layers of the retina and rods, both in photopic and scotopic conditions with electronegative ERG (Fig. 4).
3.4. Genetic Study
Regarding the genetic study, the sequencing data from the index individual showed a mean coverage of 256.3× and 95.4% of the bases had a coverage > 20x. Targeted sequencing revealed a nonsense hemizygous variant in the RS1 gene (NM_000330: c.98G>A; p.Trp33*). Sanger sequencing of exon 3 of RS1 showed that the variant co-segregated with the disease in the 3 siblings.
4. DISCUSSION
Clinical diagnosis is not easily determined in certain cases because of the wide range of phenotypes, and this can delay a diagnosis [4].
The suspected diagnosis of the index patient, before he was referred to our center, was of subsequent uveitis associated with CME. In addition, we found an atypical disease for 2 reasons: the advanced age of presentation, the average is between 18 and 24 years, although with wide ranges [5, 6], and the scattered whitish mottling seen in fundoscopy; hyperautofluorescence and hyperfluorescence were similar to that found in Retinitis Punctata Albescens (RPA), a form of atypical Retinitis Pigmentosa (RP). The clinical features of RPA are night blindness from childhood, progressive loss of visual acuity due to macular degeneration, the presence of small white deposits and patches of atrophy in the peripheral retina that contrast with the absence or scarcity of pigment deposits, and the predominant participation of the bar on the cone [7]. The association of both entities (RP and XLRS) is very unusual; previous findings have only described 1 case where the presence of spicules abroad is associated with fundoscopy and mutation in the RS1 gene in the genetic study [8].
For this reason, the use of complementary tests, electrophysiology, and genetic study is particularly useful for diagnosis. In OCT, the most characteristic findings are the presence of macular schisis (82-88%) followed by macular atrophy (11-18%) [5, 6]. Age has also been correlated with Central Macular Thickness (CMT) and with the length of the outer layer of photoreceptors (PR); so that the older, the younger, the GMC and PR [5]. Thus, it seems that the natural progression of the disease is to central macular atrophy over time, and this evolution was observed in our patients [5].
Electrophysiology is essential in the diagnosis of retinal dystrophies. The most characteristic alteration in XLRS is the presence of negative ERG, b-wave is reduced, while a-wave remains unchanged; these are the findings that we found in our patients. It is also important for differential diagnosis since the negative ERG allows us to differentiate XLRS, especially from the Goldmann-Favre syndrome, in which we would expect to find a flat ERG [9].
The definitive diagnosis was determined by a genetic study. The mutation presented in our patients (NM_000330: c.98G>A; p.Trp33*) had previously been reported as pathogenic [10].
CONCLUSION
Interestingly, the index patient manifested only macular retinoschisis without peripheral alterations. Moreover, the characteristic fundus appearance of retinitis punctata albescens, present in our family, was absent in the Chinese patient, thus reinforcing the establishment of the new genotype-phenotype correlation that we propose. The interfamilial phenotypic variability could be due to different factors and mechanisms influencing the expression of the phenotype, such as the presence of genetic modifiers. Our work contributes to expanding information regarding the genotype-phenotype correlations associated with RS1 gene mutations that are crucial for improving the diagnosis, prognosis, and clinical management of patients with inherited retinal dystrophies. We believe future investigations should be centered on developing successful gene therapy in order to offer potential treatments to our patients.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Approved by the Institutional Review Boards of the University Hospitals Virgen del Rocio and Virgen Macarena, Seville.
HUMAN AND ANIMAL RIGHTS
No Animals were used in this research. All human research procedures were followed in accordance with the ethical standards of the committee responsible for human experimentation (institutional and national), and with the Helsinki Declaration of 1975, as revised in 2013.
CONSENT FOR PUBLICATION
An informed consent form for clinical and molecular genetic studies was signed by all participants.
STANDARDS OF REPORTING
CARE guidelines have been followed.
AVAILABILITY OF DATA AND MATERIALS
The nomenclature of variants was adjusted to the Human Genome Variation Society (http://varnomen.hgvs.org/) guidelines using Mutalyzer (https://mutalyzer.nl/).
FUNDING
This study was funded by RETICS Oftared “RD 16/0008/0010”, the European Regional Development Fund (ERFD), Carlos III Institute of Health, Spanish Ministry of Economy and Competitiveness, through the projects PI15-01648 and PI15-01648 (co-funded by the European Regional Development Fund, “A way to make Europe”), CIBERER ACCI [ER16P1AC702/2017], and the Regional Ministry of Health and Families of the Autonomous Government of Andalucia [PEER-0501-2019]. The Foundation Isabel Gemio/Fundación Cajasol. The CIBERER is an initiative of the ISCIII, Spanish Ministry of Economy and Competitiveness.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENETS
Declared none.

