All published articles of this journal are available on ScienceDirect.

Necrotizing Anterior Scleritis in a Woman with Terrien’s Marginal Degeneration: A Case Report
Authors Info & Affiliations
Abstract
Purpose:
The aim of the study was to report the first case of a patient with Terrien’s Marginal Degeneration (TMD) who developed necrotizing anterior scleritis without systemic disease association, requiring systemic immunosuppressive treatment.
Case Report:
A 32-year-old female consulted for bilateral ocular burning and hyperemia. Initially, she was diagnosed with conjunctivitis and treated with topical antibiotics and corticosteroids, with mild transitory improvement but the progression of the disease. Years later, she attended the ocular immunology consultation for a second opinion where TMD with ocular inflammatory component OU was diagnosed. Seven months later, she presented with severe pain, decreased visual acuity, and photophobia in OS. At the slit-lamp examination, necrotizing anterior scleritis with a high risk of perforation in OS was observed. The patient was referred to the rheumatologist and started treatment with systemic corticosteroids and cyclophosphamide, exhibiting a clinical improvement. The patient did not meet the criteria for any systemic illness associated with scleritis, such as autoimmune diseases or vasculitis. Thus, scleritis was related to the adjacent inflammatory process associated with TMD, as an atypical presentation of this disease.
Conclusion:
Although an inflammatory type of TMD has been proposed, it is essential to follow up closely these patients and consider necrotizing anterior scleritis, a severe ocular disease that requires prompt immunosuppressive management, as a possible atypical associated presentation of this disease.
1. INTRODUCTION
Scleritis is a severe inflammatory condition characterized by edema and inflammatory cell infiltration of the sclera [1]. Scleritis is classified according to the anatomical site and characteristics of the inflammation in the anterior (diffuse, nodular, and necrotizing) and posterior (diffuse and nodular) segments of the eye. Necrotizing anterior scleritis is divided into two forms, with inflammation or without inflammation (scleroma-lacia perforans). This disorder occurs more frequently in women older than 60 years and is frequently associated with systemic inflammatory diseases [2]. Besides, Terrien’s Marginal Degeneration (TMD) is a quite rare disease that usually presents in men between 40 and 59 years of age [3]. TMD typically presents as a unilaterally or asymmetrically bilateral peripheral corneal thinning, peripheral opacity with lipid deposition, corneal neovascularization, pseudopterygium formation, and astigmatism [3].
Although scleritis has been strongly associated with systemic inflammatory and infectious diseases, and one of its most common complications is corneal involvement, such as peripheral corneal thinning, stromal keratitis, and peripheral ulcerative keratitis (PUK), the association between necrotizing scleritis and TMD has never been reported [1]. To the best of our knowledge, we present the first case of a woman with TMD who developed necrotizing anterior scleritis without systemic disease association, requiring systemic immunosuppressive treatment.
2. CASE REPORT
A 32-year-old Colombian female, without any past history, consulted for bilateral ocular burning and hyperemia without discharge. At first, she was diagnosed with infectious conjunctivitis and treated with topical antibiotics and corticosteroids, with mild transitory improvement but disease’s progression. A cornea specialist, who suspected an undifferentiated autoimmune disease, later evaluated the patient. Thus, she was referred to the rheumatologist, who asked first for an autoimmune profile (Table 1), ruled out infectious causes (Herpes simplex, Syphilis, and Tuberculosis), and initiated immunomodulatory therapy with deflazacort 30mg bid for 10 days, methotrexate 15 g per week, and folic acid without a specific systemic associated diagnosis.
Years later, at 41 years of age and after 9 years of symptomatology, the patient attended the ocular immunology consultation for a second opinion. The patient’s external examination showed bilateral photophobia, pseudoptosis, and the best-corrected visual acuity (BCVA) was 20/20-1 OD and 20/20-2 OS. Slit-lamp examination in OD revealed mild telangiectasias on the lid margin, mild meibomian gland dysfunction, severe conjunctival 360-degree hyperemia, major ciliary injection, and generalized vascular tortuosity. The cornea had a 360-degree peripheral opacity with whitish infiltrate at the edges, greater in the superior nasal sector, with neovascularization in the superior and inferior periphery, and with corneal thinning at the lower region not staining with fluorescein. OS findings were equal to OD, except for an additional whitish lesion in the nasal region of approximately 4 mm, which crossed the limbal margin (Fig. 1). Intraocular pressure was 17/15 mmHg. Ocular fundus examination was normal OU. According to these clinical findings, inflammatory TMD and secondary pseudopterygium OS were diagnosed. She was treated with topical prednisolone acetate 1%, lubricant ophthalmic gel, and topical sodic diclofenac 0.1%. At the second follow-up, mild improvement was evidenced; thus, topic prednisolone was changed to loteprednol 0.5%, and the rest of the treatment was continued.
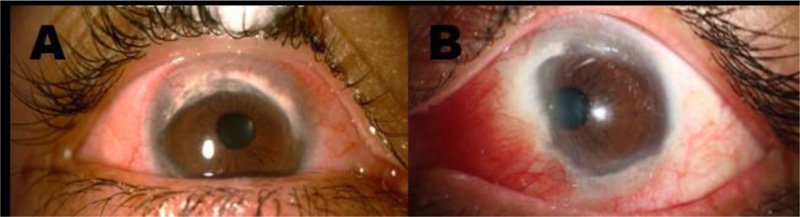
Slit-lamp examination A. OD: Conjunctival 360-degree hyperemia, ciliary injection, and generalized vascular tortuosity. The cornea shows a 360-degree peripheral opacity with whitish infiltrate at the edges, greater in the superonasal quadrant, with neovascularization in the superior and inferior periphery, and corneal thinning at the lower region. B. OS: Severe conjunctival hyperemia in the nasal quadrant. The cornea shows a 360-degree peripheral opacity with whitish infiltrate at the edges, greater in the superior sector with neovascularization in the superior and inferior periphery, and corneal thinning at the lower region. Additionally, a whitish/pink lesion of approximately 4 mm, crossing the limbal margin, compatible with a pseudopterygium, is observed in the nasal region.
| Diagnostic Test | Previous Results | Last Results (2020) |
|---|---|---|
| C-Reactive Protein | <0,4 mg/dl (negative) | negative |
| Erythrocyte sedimentation rate | 3 ml/h (normal) | 24 ml/h (normal) |
| ANAS antibodies | 1: 160 to 1: 320 (speckled pattern) between 2012-2019 | |
| ANCAS (IFI & ELISA) antibodies | negative | |
| ACPA antibodies | 5.0 EU/mL(negative) | |
| Rheumatoid Factor | 1.1 UI/ml (negative) | |
| Anti-ENA antibodies | negative | |
| Anti-DNA antibodies | negative | |
| Anti-MPO antibodies | Negative | 5.6 U/mL (positive) - (05/10/20) reference value 5 |
| 4.4 U/mL (negative) - (08/10/20) | ||
| Anti- PR3 | Negative | 1.4 U/mL (negative) |
| Complement (C3-C4) | C3: 123 mg/dL (normal) C4: 23.7 mg/dL (normal) |
C3: 143 mg/dL (normal) C4: 24.3 mg/dL (normal) |
| Anti-CCP IgG | 1.4 - 4.4 5 U/ml (negative) | |
| Anti-Smith antibodies | 2.51 - 6.8 U/mL (negative) | 1 U/mL (negative) |
| Anti-RNP antibodies | 2.17 - 18.0 U/mL (negative) | 0.5 U/mL (negative) |
| Anti-Ro antibodies | 2.1 - 20.5 U/mL (negative) | 2 U/mL (negative) |
| Anti-La antibodies | 1.69 - 9.8 U/mL (negative) | 7.8 U/mL (negative) |
| HLA B27 | Negative | |
| Salivary gland biopsy | ● Preserved architecture, acinar and ductal structures without atypia or presence of inflammation. ● Diagnosis: Essentially normal (Grade 0 of the Chisholm and Manson classification). |
|
Seven months later, she presented severe pain, decreased BCVA, and photophobia in OS. At the slit-lamp examination, a rounded severe scleral thinning area in the OS nasal perilimbal zone with uveal tissue translucence (Fig. 2) was observed, leading to a diagnosis of necrotizing anterior scleritis with a high risk of perforation. Given this finding, the patient was referred to the rheumatologist and was admitted to the hospital. She was treated with three methylprednisolone pulses (1gr IV q.d) and a single dose of cyclophosphamide (850mg IV). In addition, a new complete diagnostic test panel was performed, revealing slightly positive anti-MPO antibodies as a unique finding. However, antibodies were repeated exhibiting a negative result, thus inflammatory systemic diseases were ruled out (Table 1). The patient improved with this systemic management, and the treatment was continued with oral prednisolone (60 mg/day) and five doses of cyclophosphamide (1gr IV monthly). The patient did not meet criteria for any systemic disease and was considered as a presumptive first vasculitis manifestation without extraocular involvement still developed. Nevertheless, necrotizing scleritis associated with TMD was also considered.
After two weeks, the patient was followed-up by the ophthalmologist, and she was found to have a substantial improvement, so corticosteroid tapering was started. In the most recent follow-up, 6 months after the initial evaluation, BCVA was 20/25 OD and 20/50 OS. At the slit-lamp examination, OU had TMD features without hyperemia and with a marked scleritis improvement. Methotrexate 15mg per week was initiated as a chronic corticoid sparing agent due to the severity of the clinical picture.
3. DISCUSSION
Necrotizing scleritis represents 4% of all scleritis cases, is more frequent in women older than 60 years, and has unilateral involvement in 60% of cases [4]. This coincides with our patient’s sex and unilateral involvement, however, our patient was younger.
Necrotizing scleritis diagnosis is based on clinical findings and is frequently associated with systemic inflammatory diseases, especially with connective tissue or vasculitis diseases [4], which in our case could not be documented.
On the other hand, TMD is a rare disease, with no reported incidence or prevalence. It commonly occurs in males around 40 years of age [3]. TMD is characterized by thinning and furrow formation of the peripheral cornea, superficial vascularization with radial trajectory, tortuosity and occasional circumferential appearance in the thinned area, lipid deposition at the leading edge of the affected area, and intact corneal epithelium without apparent inflammatory signs or symptoms [3]. It usually has bilateral involvement and initiates at the superior or superonasal cornea, generating peripheral ectasia and sometimes a pseudopterygium [4, 5]. All of these findings coincide with our patient’s disease presentation.
In TMD, it is important to rule out differential diagnoses, such as PUK, pellucid marginal degeneration (PMD), and Mooren’s ulcer [3]. In our case, PUK was discarded because it generally does not present 360 degrees’ involvement, has epithelium loss, and is associated with systemic vasculitis; PMD was also discarded, as it does not present lipid deposition or neovascularization. And finally, Mooren’s ulcer was discarded because it is characterized by corneal thinning with an “overhanging” edge and disruption of the epithelium, which was not present in our patient [3].
Regarding the association between TMD and necrotizing scleritis, Iwamoto et al. [6] and Austin et al. [7] have proposed an inflammatory type of TMD, in which patients present typical signs of TMD associated with recurrent symptoms of ocular surface inflammation and episcleritis or superficial scleritis. However, in our case, the patient presented a more severe type of scleritis. To the best of our knowledge, we present the first case of a woman with TMD associated with necrotizing anterior scleritis.
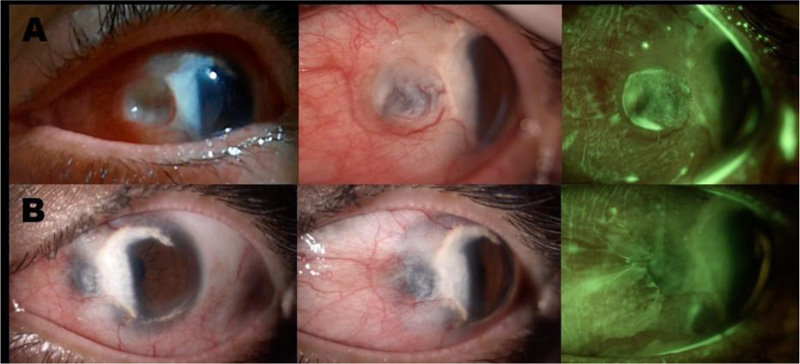
Slit-lamp examination A. Findings during the episode of active necrotizing anterior scleritis. OS: Conjunctival hyperemia, vascular tortuosity, excavated lesion in a rounded nummular shape, secondary to severe scleral thinning with uveal exposure. Fluorescein staining shows a zone of atrophy with tear accumulation, without perforation or aqueous humor leakage. B. Improvement of scleral thinning with the persistence of uveal tissue visualization. The cornea shows TMD features with a whitish lesion in the temporal region (from 5 to 1 o’clock) which crosses part of the visual axis. Additionally, in the fluorescein staining photograph, improvement of the scleral lesion after treatment is evidenced.
Scleritis is commonly associated with systemic diseases (45-95%) [4], especially with rheumatoid arthritis, systemic lupus erythematosus, and vasculitic diseases, such as granulomatosis with polyangiitis [8]. Likewise, a few cases have been reported in which TMD is associated with systemic diseases, such as rheumatoid arthritis [9], idiopathic juvenile arthritis [10], erythema elevatum diutinum [11], and systemic vasculitis [12]. Although in our case, the patient had anti-MPO antibodies positive once and systemic diseases were suspected, especially microscopic polyangiitis, the patient was fully studied by the rheumatology service and did not meet the criteria of any disease according to ACR criteria and the Chapel Hill definition [13].
Considering the borderline anti-MPO positivity presented in our patient, it is important to remark that we did not discard a possible latent autoimmune disease with initial ocular involvement [14]. However, even if it is the case, the association between TMD and necrotizing anterior scleritis is still rare, and it is important to continue studying the pathophysiological events of TMD and its possible relationship with inflammatory eye diseases.
CONCLUSION
Finally, considering that necrotizing anterior scleritis is a severe disease that could lead to ocular perforation, we recommend performing adequate and strict follow-ups on patients with inflammatory TMD. Furthermore, in case of suspicion of a similar situation to the one presented in this report, the specialist must consider vigorous immunosuppressive management and immediately refer to the uvea and rheumatology specialist consultation.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
This study adhered to the ethical principles for human research established by the Helsinki Declaration, the Belmont Report, and Colombian Resolution 008430 from 1993.
CONSENT FOR PUBLICATION
In the consent form, the patient has given her consent for images and the clinical information to be reported in the journal. The patient understands that her name and initials will not be published, and due effort will be made to conceal her identity.
STANDARDS OF REPORTING
CARE guidelines and methodologies were followed for this study.
AVAILABILITY OF DATA AND MATERIALS
The data generated during the current study are available from the corresponding author on reasonable request.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
The authors thank the patient for granting permission to publish this information and Universidad del Rosario for financing the publication charges of this article.
Authorship: All authors attest that they meet the current ICMJE criteria for Authorship.

