All published articles of this journal are available on ScienceDirect.

Identification of a PROM1 mutation in a Spanish family with inherited retinal dystrophies
Abstract
Background:
We report a Spanish family, comprising an affected mother and daughter, respectively diagnosed with retinitis pigmentosa and Stargardt-like macular dystrophy, in whom we identified a PROM1 mutation.
Methods:
A custom gene panel consisting of 119 inherited retinal dystrophies (IRD)-genes was applied in the two affected individuals of this family and sequenced using the Illumina´s NextSeq500 platform.
Results:
The analysis of the resulting data allowed us to identify the pathogenic PROM1 mutation c.1117C>T (p.Arg373Cys) as the primary cause of the disease in both patients. No additional variants contributing to the extent of retinal dysfunction were detected.
Conclusion:
The variable expressivity of the detected PROM1 mutation is the most likely responsible for the intrafamilial phenotypic variability observed in this family. Screening of this mutation should be considered in patients with compatible clinical manifestations, especially when accompanied by an autosomal dominant family history.
1. INTRODUCTION
Prominin-1 (PROM1), also known as CD133, is a cholesterol-binding protein that is selectively associated with plasma membrane protrusions. In the visual system, PROM1 is concentrated in the photoreceptor outer segment disc membranes and its dysfunction may cause serious visual impairment. Mutations in PROM1 are associated with hereditary macular and cone‒rod dystrophies (CRDs), such as Stargardt-like and bulls’ eye macular dystrophies (BEM), retinitis pigmentosa, and cone‒rod dystrophy [1].
CRDs include diverse inherited retinal diseases with retinal alterations involving the central retina. Macular dystrophies are the leading cause of visual impairment leading to irreversible blindness in the development world. Autosomal dominant Stargardt-like macular dystrophy is one of the early onset macular dystrophies characterized by a progressive loss of central vision and atrophy of the retinal pigment epithelium (RPE). Clinical features include progressive loss of central vision, minimal to no color vision defects, and no significant changes in electroretinogram (ERG) [2].
We here report a Spanish family, comprising an affected mother and daughter, respectively, diagnosed with retinitis pigmentosa and Stargardt-like macular dystrophy, in whom we identified a PROM1 mutation.
2. MATERIALS AND METHODS
The proband was a 37-year-old woman diagnosed with Stargardt-like macular dystrophy at the age of 35 years (Patient 1). She complained of central visual disturbance. Her mother had been diagnosed with retinitis pigmentosa. This family history of visual impairment suggested an autosomal dominant inheritance pattern.
An electroretinogram (ERG) showed reduced amplitudes and delayed cone responses, with rod responses within normal limits. These are the classic ERG findings of rod dystrophies and maculopathies, such as Type 2 Stargardt disease.
The mother of the proband was a 57-year-old woman diagnosed with retinitis pigmentosa (Patient 2). She complained of progressive loss of central vision. Her BCVA was 20/40 in the right eye and 20/20 in the left eye. The ERG pattern shows abnormalities of both cone and rod responses.
Genomic DNA of the patients was isolated from peripheral blood using ChemagicTM 360. DNA samples were subjected to targeted sequencing using the SeqCap EZ Choice Enrichment kit and the NextSeq500 instrument. The custom panel covered all exons plus 10 bp of intronic flanking regions of 119 inherited retinal dystrophy (IRD)-associated genes (Table S1). A DNA library was generated according to the manufacturer’s protocol [3] and data analysis was conducted by our validated pipeline. To prevent the loss of second-site modifiers, non-coding variants not shared between the two affected individuals were also considered. The BEDtools coverage command and subsequent normalization were used to identify copy number variations (CNVs).
3. RESULTS
Patient 1 underwent a complete ophthalmic examination. Her best corrected visual acuity (BCVA, Snellen) was 20/20 bilaterally. Slit-lamp biomicroscopic examination showed no remarkable findings in the anterior segments.
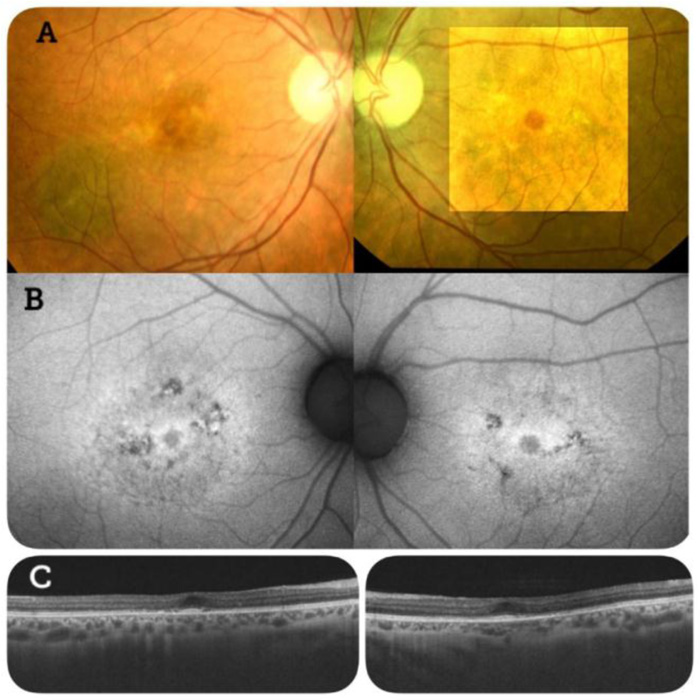
Fundus examination showed bilateral atrophic macular lesions with yellowish spots at the perifoveal region. Fundus autofluorescence showed hypo-autofluorescent macular lesions surrounded by hyper-autofluorescence. Optical coherence tomography (OCT) scans were performed in both eyes. These revealed RPE atrophy and photoreceptor layer defects (Fig. 1).
An electroretinogram (ERG) showed reduced amplitudes and delayed cone responses, with rod responses within normal limits. These are the classic ERG findings of rod dystrophies and maculopathies, such as Type 2 Stargardt disease.
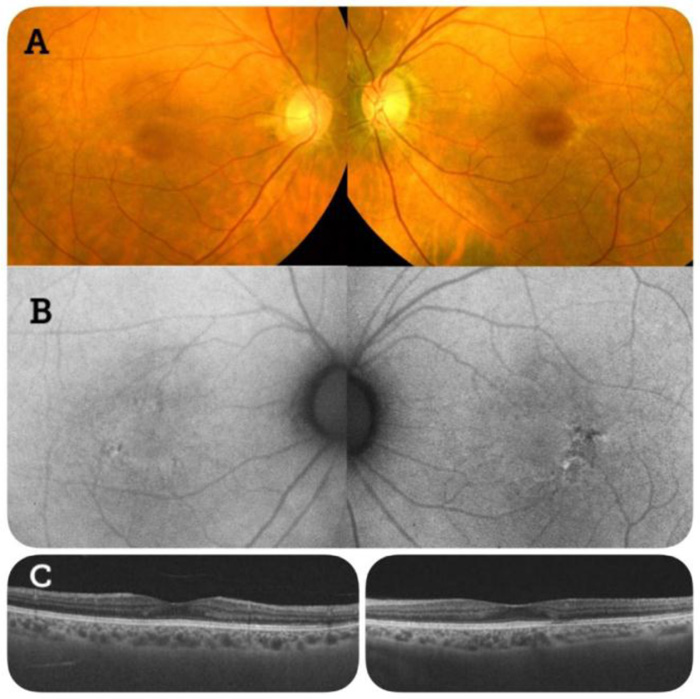
Data obtained from fundoscopy and fundus autofluorescence of patient 2 were compatible with a rod‒cone dystrophy (Fig. 2).
Sequencing data from both affected individuals resulted, on average, in 83.35% reads mapping on target and a mean coverage of 290.1×, with 98% of bases covered > 20×. Prioritization criteria allowed us to analyze a mean of 10 variants per sequenced sample. Among them, only one missense mutation in PROM1 (c.1117C>T; p.Arg373Cys) could be considered disease-causative. Both patients harbored this variant in a heterozygous state with a specific depth greater than 200×. This mutation had previously been reported to be pathogenic both in public databases and in the literature [4]. No modifier alleles or CNVs were identified.
PROM1 is located in the photoreceptor outer segment disc membranes, where it plays a structural role. The deletion of PROM1 leads to ineffective autophagosomal‒lysosomal‒phagocytic pathways, which can impair clearance of shed photoreceptor outer segments and lead to accumulation of damaged organelles and protein aggregates within the cell, including lipofuscin-like debris, linking to the pathogenesis of macular disease [1].
4. DISCUSSION
The mutation c.1117C>T is relatively recurrent among patients with autosomal dominant IRD [5]. Although BEMs are the most common form of disease related to this mutation [4], additional phenotypes varying both within and among families have also been reported [6]. This large intragenic variability can be attributed to the extensive phenotypic heterogeneity of monogenic diseases and/or to the role of non-allelic variations in shaping genotype‒phenotype correlation. Nevertheless, we identified no genetic modifiers that could justify the intrafamilial phenotypic variability observed in the two affected family members. Therefore, these results support the previously assigned variable expressivity for the PROM1 p.Arg373Cys mutation. However, given the ubiquitous nature of genetic modifying factors, their existence in non-analyzed regions cannot be ruled out and further large-scale studies are needed to ascertain that the phenotypic variability is due to the mutation itself. Confirming that variable expressivity of some causal variants could underlie the phenotypic variability would allow improvement of clinical and genetic management of patients and could guide the search for certain pathogenic mutations when particular phenotypic hallmarks are observed.
Mutations in PROM1 associated with hereditary retinal diseases have been previously described. The most recent case described in the literature of this mutation was reported by Kim et al. in a Korean patient with Stargardt-like dystrophy [7]. Moreover, Michaelides et al. identified the related chromosomal locus in 11 members of a five-generation British family with a dominantly inherited macular dystrophy [8]. Recently, Liang et al. identified two novel deletion mutations in PROM1 associated with macular and rod‒cone dystrophy in a Han Chinese family [9]. In 2011, Arrigoni et al. described the role of the same PROM1 mutation as identified in our cases in three members of a family with autosomal dominant macular degeneration [10].
CONCLUSION
In conclusion, we here presented two related Spanish cases with different IRDs in whom a single mutation in PROM1 was found by targeted sequencing, suggesting that the causative mutation is also responsible for the observed clinical variability.
Nevertheless, since both individuals carried the same mutation, we cannot exclude age as a factor explaining the variable phenotype when considering the presentation of daughter versus mother.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was approved by the Institutional Review Boards of the Hospitals Virgen del Rocio and Virgen Macarena, Seville (I15_01648 and CTS1664).
HUMAN AND ANIMAL RIGHTS
No animals were used in this research. All human research procedures were followed in accordance with the ethical standards of the committee responsible for human experimentation (institutional and national), and with the Helsinki Declaration of 1975, as revised in 2013.
CONSENT FOR PUBLICATION
Informed consent form was signed from all participants for clinical and molecular genetic studies.
STANDARDS OF REPORTING
CARE guidelines and methodologies were followed for this study.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors have no financial interests related to any of the materials or equipment used in this study.
ACKNOWLEDGEMENTS
Declared none.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s website along with the published article.

