All published articles of this journal are available on ScienceDirect.

Optic Neuritis, its Differential Diagnosis and Management
Abstract
The aim of this review is to summarize the latest information about optic neuritis, its differential diagnosis and management. Optic Neuritis (ON) is defined as inflammation of the optic nerve, which is mostly idiopathic. However it can be associated with variable causes (demyelinating lesions, autoimmune disorders, infectious and inflammatory conditions). Out of these, multiple sclerosis (MS) is the most common cause of demyelinating ON. ON occurs due to inflammatory processes which lead to activation of T-cells that can cross the blood brain barrier and cause hypersensitivity reaction to neuronal structures. For unknown reasons, ON mostly occurs in adult women and people who live in high latitude. The clinical diagnosis of ON consists of the classic triad of visual loss, periocular pain and dyschromatopsia which requires careful ophthalmic, neurologic and systemic examinations to distinguish between typical and atypical ON. ON in neuromyelitis optica (NMO) is initially misdiagnosed as ON in MS or other conditions such as Anterior Ischemic Optic Neuropathy (AION) and Leber’s disease. Therefore, differential diagnosis is necessary to make a proper treatment plan. According to Optic Neuritis Treatment Trial (ONTT) the first line of treatment is intravenous methylprednisolone with faster recovery and less chance of recurrence of ON and conversion to MS. However oral prednisolone alone is contraindicated due to increased risk of a second episode. Controlled High-Risk Subjects Avonex® Multiple Sclerosis Prevention Study “CHAMPS”, Betaferon in Newly Emerging Multiple Sclerosis for Initial Treatment “BENEFIT” and Early Treatment of MS study “ETOMS” have reported that treatment with interferon β-1a,b results in reduced risk of MS and MRI characteristics of ON. Contrast sensitivity, color vision and visual field are the parameters which remain impaired mostly even after good recovery of visual acuity.
INTRODUCTION
The term optic neuritis (ON) refers to inflammation of the optic nerve due to many causes, indicated by sub-acute unilateral painful visual loss mostly in a young healthy female and by excluding glaucoma, ON is the most common optic neuropathy in persons under 50 years coming to general ophthalmic practice. It is the earliest clinical symptom in about 20% of cases of MS [1-5].
The gold standard treatment for ON is based on the Optic Neuritis Treatment Trial (ONTT) which was undertaken in order to determine the efficacy of corticosteroids and to permit long-term analyses [1,6,7]. In the ONTT, 15 clinical centers in the United States registered 457 patients between July 1, 1988 and June 30, 1991 with the following criteria: the presence of acute unilateral optic neuritis with visual symptoms for 8 days or less, age between18 and 45 years, no previous history of ON in the affected eye, no evidence of systemic disease other than MS that might be associated with the ON, and no previous treatment with corticosteroids for MS or ON [1,6,7]. This article reviews available studies published in English and Spanish regarding optic neuritis, its differential diagnosis and management.
AETIOLOGY
ON is mainly idiopathic in nature; though, it could be related to demyelinating lesions (e.g. MS [4,8-10] neuromyelitis optica (NMO) [4,8,9]) or other less common etiologies such as autoimmune disease (e.g. sarcoidosis [1,4,9], systemic lupus erythematosus (SLE) [4,9]), infectious and para-infectious causes (e.g. syphilis [1,4,9,11], tuberculosis [4,9,11]), inflammatory and post vaccination immunological responses [4] (e.g. sinusitis [1], and vaccinations against measles and rubella [12]). (Table 1)
Aetiology of Optic Neuritis
| Demylinating lesions | Multiple Sclerosis (MS) [4,8-10], Neuromyelitis optica [4,8,9], Shilder’s disease, Encephalitis periaxialis concentrica [4] (out of which MS is the most common cause [4]) |
| Autoimmune Disease | Sarcoidosis [1,4,9], systemic lupus erythematosus (SLE) [4,9], Sjögren’s syndrome (SS), Behchet’s disease [9] |
| Infectious/para-infectious | Herpes zoster [9], lyme disease [1,4,9], syphilis [1,4,9,11], tuberculosis, dengue [11,13], mumps, varicella zoster [4,11], toxoplasmosis [4,9,11], measles [4,11,12], leptospirosis, chickungunya, west nile [11], adenovirus, brucellosis, coxsackievirus, cat scratch disease, β-hemolytic streptococcal infection, meningococcal infection, typhoid fever, whipple’s disease [4] |
| Inflammatory/post vaccination | sinusitis [1], vaccinations against tuberculosis, hepatitis B, rabies, tetanus, meningitis, anthrax, measles, rubella & influenza [12] |
Features of Typical ON in Adults
| • Acute to sub-acute onset [2,8] promoting over a several hours 6 to 2 weeks [2,9,10] |
| • Young adult patient with peak manifestation between 15-50 years of age [2,3,9] |
| • Females > males [2,3,9] |
| • Periocular pain (90%) [2,5,16-18], especially with eye movement [2,3,8-10,18] |
| • Unilateral loss of visual acuity [2,4,8-10,18] variable in severity (from 20/20 in 10.5% to no light perception in 3.1% [4,5,16]), or may be bilateral usually in children often associated with a post or para infectious demyelination [2,4,8,10] |
| • Reduced contrast sensitivity [1,8,9,18] |
| • Uhthoff’s phenomenon (Exercise or heat-induced deterioration of visual symptoms) [2,5,8,18] |
| • Pulfrich phenomenon (misperception of the direction of movement of an object) [2,5,8,18] |
| • Ipsilateral relative afferent pupillary defect (RAPD). lack of the defect suggests a preexisting or concurrent optic neuropathy in the fellow eye [2,4,8-10,18] |
| • Normal (65%) or swollen (35%) (more common in children) optic nerve head [2,5,9,10,18] |
| • Possibility of mild uveitis [9,18] and retinal periphlebitis [2,9,18] |
| • Visual field defect [2,4,10], any type [9,18]; ranging from commonly seen diffuse depression and central or centrocecal scotoma [2,5,8,16], to rarely seen quadrantic [2,4,16] and altitudinal defects [4] |
| • Spontaneous visual improvement in >90% [2,3,9,18] |
| • No deterioration in vision after steroids discontinuation [2,9] |
| • Pallor of the optic disc [2,11] |
| • Previous history of ON or MS [9,18] |
| • Reduction in vision in bright light [2] |
| • Phosphenes or photopsias (spontaneous flashes of light in vision) provoked by eye movement [8,18] |
| • Dyschromatopsia [2,8-10,18] (any type) [2,7]* |
Features of Atypical ON in Adults
| • Age > 50 or < 12 years [2,9] |
| • Simultaneous or sequential bilateral ON [2,4,9] |
| • light perception) which progress for > 2 weeks from onset [1,2,4,9] |
| • Painless/painful/persistent pain > 2 weeks [2,4,9] |
| • Abnormal ocular findings: |
| • Absence of any visual recovery within 3-5 weeks [2,4,9] or continued exacerbation in visual function [2] |
| • Lower risk of developing MS [4] |
| • Manifestation of systemic diseases other than MS [2,9] |
| • Deterioration in vision after steroids discontinuation [2,9] |
| • Family history [2] |
| • Previous history of neoplasia [2,9] |
| • Optic atrophy lacking history of ON or MS [9] |
Differential Diagnosis of NMO vs MS
| Features | Neuromyelitis Optica | Multiple Sclerosis | |
|---|---|---|---|
| Attacks are bilateral | Usually [8,9] | Rarely [8] | |
| Visual loss severity | More, with less improvement [8,10,14,22] | Less, with more improvement [9,22] | |
| White matter lesions on brain MRI | Rarely and usually resolving [8,9] | Usually [8,22] | |
| Transverse myelitis | TM in spinal MRI often spanning ≥3 spinal cord segments (in 20%) [2,8,9,22] | Rarely [8] | |
| Clinical involvement beyond spinal cord and optic nerve | Rarely [8] | Usually [8] | |
| Tissue destruction and cavitations | More than MS [8] | Less than NMO [8] | |
| CSF Analysis | Oligoclonal bands | Rarely [2,8,9] | Frequently [8,22] |
| Protein contents | Higher than MS [8] | Lower than NMO [8] | |
| Treatment | DMDs | Ineffective even worsening [8,9] | Effective [8,9,23] |
| Immunosuppressive (corticosteroids) | First line of treatment [8,9] | First line of treatment [24] | |
PATHOPHYSIOLOGY
The pathogenesis of optic neuritis is not well understood. It is likely due to some inflammatory process which leads to delayed type IV hypersensitivity reaction induced by released cytokines and other inflammatory mediators from activated peripheral T-cells which can cross the blood brain barrier and cause destruction of myelin, neural cell death and axonal degeneration.
Latest technologies such as optical coherence tomography (OCT) suggest involvement of axons (gray matter) in addition to myelin sheath (white matter) in this process [2,5,8,14].
Permanent visual loss (40%to 60%) and visual deficit in ON is a result of axonal loss in the optic nerve and retina and corresponding retinal nerve fiber layer (RNFL) thinning, in addition to conduction block caused by demyelination of the optic nerve [8,14,15].
CLINICAL FEATURES AND DIAGNOSIS
Based on the location of involvement, ON can be categorized as: 1. retrobulbar neuritis (2/3 of cases) with normal optic disc appearance; 2. papillitis with swollen disc; 3. perineuritis, which involves the optic nerve sheath while the optic disc may or may not be swollen;4. neuroretinitis with optic disc oedema and macular star exudates.
Retrobulbar neuritis and papillitis are mainly associated with MS while perineuritis and neuroretinitis are more often associated with infectious or inflammatory pathologies [1,2,4,8,16].
Based on its clinical features, ON can be classified as atypical or typical which is present without any manifestation of systemic disease and may occur either as a clinically isolated syndrome or in association with MS (Tables 2 and 3) [2,8,9].
The classic triad for diagnosis of ON is visual loss, periocular pain and dyschromatopsia [2,5,9]. Optic neuritis can have long-lasting effects on visual motion processing regardless of temporary effects on visual form processing. In the acute phase there is a much greater effect on motion processing than form processing [20].
ON Associated with NMO (Devic’s disease) & MS
NMO is an acute inflammatory demyelinating disease mainly involving the optic nerves and spinal cord. Optic neuritis in NMO and MS are nearly identical in their initial presentation. However, demyelinating NMO is more violent and devastating than MS, hence its correct diagnosis is very important (Table 4). In more than 85% of patients with NMO, attack recurs in the form of ON, transverse myelitis (TM), or both, resulting in around 50% of cases in paralysis or blindness within 5 years. Sometimes patients with TM in the cervical spine experience respiratory failure and even death. Serum NMO-IgG, a biomarker of NMO, is detected in 70% of patients, and targets the water channel protein aquaporin-4. The diagnosis of NMO requires two absolute criteria and two of the three supportive criteria (Table 5) [2,8,9,14,21].
MS is a demyelinating disease disseminated in time and space i.e., the incidence of a second clinical episode at a distinct site in the central nervous system (CNS). Absence of common deficit along with lack of abnormal findings in MRI or cerebrospinal fluid (CSF) exclude a diagnosis of MS [12,18,21].
According to International Panel on Multiple Sclerosis Diagnosis, MRI visualized dissemination of CNS lesions in time and space is sufficient for the diagnosis of MS even before occurrence of clinical symptoms [18].
PREVALENCE
Patients with acute demyelinating ON are typically healthy young adults. Female preponderance in observed, with a ratio of approximately 3:1. For reasons still unclear, the incidence of MS associated with ON is highest in people living at higher latitudes (e.g.in northern USA, northern and western Europe; New Zealand and southern Australasia) and reduce significantly closer to the equator. Studies have reported a correlation between reduction in vitamin D (25-hydroxyvitamin D) level and increased risk of developing/relapsing MS. Therefore lower intensity and lesser exposure to sunlight at high latitude may be an explanation for epidemiological variations of MS. The annual incidence of ON is estimated at 5 per 100,000. ON is seen more commonly in Caucasians, and quite rarely in black populations. Incidence of ON is eight times higher in white northern Europeans than blacks and Asians. In Asia, ON is proportionately more common, relative to the incidence of MS in the USA or western Europe and is less frequent in south America and Mediterranean region but newer studies has reported an increasing prevalence in the last few decades. Studies have shown that peoples who migrate before puberty get the incidence of MS in the region to which they migrate. So, a connection exists between ethnicity and environment [1-4,8,9,25-28].
DIFFERENTIAL DIAGNOSIS
Various forms of optic neuropathy mimic ON, resulting in misdiagnosis. These include AION (Anterior ischemic optic neuropathy) and LHON (leber’s hereditary optic neuropathy), which most closely resemble ON. Toxic/metabolic causes and compressive optic neuropathies should also be considered in the differential diagnosis of ON [2,5,23].
- Anterior Ischemic Optic Neuropathy (AION) which occurs in individuals over 50 years of age and equally in males and females, is characterized by unilateral sudden painless (pain is present in <10% of patients; accompanied by headache in patients with temporal arteritis) loss of vision, varying from visual acuity of better than 6/6 to no light perception with impairment of color vision and altitudinal visual field defect. Crowded optic disc appears swollen with sectorial haemorrhage and small or no cup in the fellow eye. Most patients with ischemic optic neuropathy have hypertension, hypercholesterolemia, diabetes mellitus, obstructive sleep apnea, or other vascular risk factors. AION has two major subtypes. Non-arteritic AION (NA-AION) is the most common form. The visual loss in this subtype is considered to be as a result of insufficient blood supply to the optic nerve head. ON has a better prognosis than NA-AION. Arteritic AION (A-AION) is more common in females, starting with massive loss of vision, usually due to giant cell arteritis, and has a significant relationship with polymyalgia rheumatica. Patients may have jaw claudication, proximal myalgia and arthralgia, scalp tenderness, headache, fatigue, and a significantly increased erythrocyte sedimentation rate and C-reactive protein level. Amaurosis fugax is a threatening sign of impending arteritic AION. As compared to NA-AION the vision loss is more severe and the optic disc is pale. Temporal artery biopsy is the gold standard for diagnosis of AION [5,10,17,23,29,30].
- Leber’s Hereditary Optic Neuropathy (LHON) involves sub-acute and painless visual loss with central scotoma and poor color vision with sequential involvement of both eyes over a period of weeks to months. This disorder predominantly affects young men (80%–90%) and is inherited from maternal mitochondrial DNA. Funduscopic examination mainly shows circumpapillary telangiectasia, while about 1/3 of patients primarily have a normal disc appearance. Fat suppressed orbital MRI usually shows enhancement of the optic nerve in ON, but not so in AION or LHON [5,10,23].
Toxins closely associated with optic neuropathy include carbon monoxide, ethylene glycol, perchloroethylene, methanol, and tobacco. Drugs associated with optic neuropathy are ethambutol, clioquinol, isoniazid, amiodarone, linezolid, methotrexate, sildenafil, oxymetazoline, and infliximab. Moreover, various chemotherapeutic agents are identified to cause optic atrophy, including vincristine, cisplatin, carboplatin and paclitaxel. Nutritional deficiencies such as vitamin B12 in poor countries have a significant role in the endemic optic neuropathy which deteriorates by tobacco use [2,23].
Compressive optic neuropathies may be caused by sinus mucocoeles, arterial aneurism, tumors, mass lesions, thyroid eye disease or other orbital processes. Brain and orbital MRI confirms or exclude diagnoses of compressive optic neuropathy [2,16,18,23].
CLINICAL EXAMINATION
The clinical features of the patients specify the type of examination required. Generally complete ophthalmic, neurologic and systemic examinations should be performed for diagnosis of ON [4,31].
Ophthalmic examinations including slit lamp examination and pupillary reactions (RAPD) in unilateral or bilateral asymmetric conditions allows a quantitative measurement of whether the optic neuropathy is stable, improving, or worsening [1,31,32]. Aided visual acuity (V.A) is measured for near vision by near vision plates and for distance vision by ETDRS (early treatment diabetic retinopathy chart) or retro-illuminated Bailey-Lovie chart at a distance of 4m and Snellen at 6m [31,32]. Those unable to read any letters at one meter are further examined by counting fingers, identifying hand movements or perceiving light [7]. Color vision, where V.A and central visual function allows, can be recorded using FM-100 hue test (Farnsworth Munsell 100) or Ishihara pseudoisochromatic color vision plates [1,7,9]. Contrast sensitivity can be recorded using Pelli-Robson charts at a distance of 1m [7,31] or Cambridge low contrast gratings [32]. Low-contrast letter acuity (Sloan charts) and contrast sensitivity (Pelli–Robson chart) show a strong relationship with brain MRI and RNFL thickness, by OCT [2]. Visual field determination, where aided V.A permits, recorded for both eyes by Goldmann perimeter to evaluate peripheral visual field and Humphrey field analyzer to evaluate central 30 degrees [7,31,32]. Fluorescein angiography and electroretinography (ERG) is done in case of retinal diseases [4]. Optical Coherence Tomography (OCT) is used to measure the thickness of retinal tissues which are thinned in the affected eye by ON [10,14,25,33,34]. Reduction in RNFL thickness correlates with visual acuity, visual field, color vision, contrast sensitivity and visual evoked potential (VEP) amplitude [33,34].
Neurological examinations including orbital and brain MRI is performed with or without gadolinium (Gd) preferably within two weeks after the onset of symptoms [9,10,32]. Contrast enhancement of the optic nerve is a sensitive finding in acute ON but does not correlate with the degree of visual recovery [8,16] (Fig. 1A).
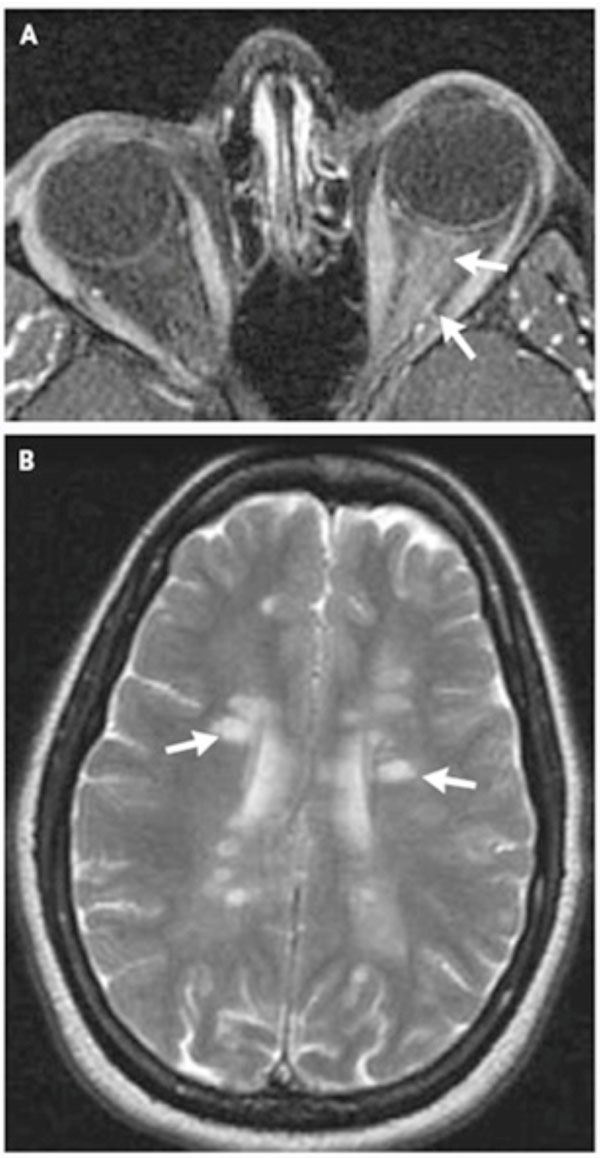
(A, B) MRI of brain and orbits in pateint with acute demyelinating optic neuritis. Reprinted by permission from [N Engl J Med 2006; 354: 1273-80. Copyright © 2006 Massachusetts Medical Society].
In patients with atypical presentation, brain and orbital MRI with Gd is compulsory and in typical ON this procedure not only allows confirmation of the diagnosis but is used as a prognosticator for developing clinically definite MS (CDMS) [1,2]. Signal abnormalities are depicted by their size (3 or <3 mm), location (periventricular or non-periventricular), and shape (ovoid or non-ovoid). No signal abnormality is categorized as grade 0; one or more focal signal abnormalities, all of which are either smaller than 3 mm or non-periventricular and non-ovoid, as grade I; one periventricular or ovoid signal abnormality at least 3 mm in size, as grade 2; two such abnormalities as grade 3; and three or more such abnormalities as grade 4 [35] (Fig. 1B). CT scan with contrast is done when MRI is not possible [2,9].
Functional MRI (fMRI) has shown evidence of changed pattern of activation of visual and some non-visual cortical areas following ON [14]. VEP may be helpful in diagnosis of subclinical cases with presentation of dyschromatopsia and optic disc pallor, contralateral subclinical cases of ON and suspected acute demyelinating ON. Abnormal results such as increased latencies and reduced amplitudes of waveform are compatible with demyelination in the afferent visual pathways and are seen in more than 65% of patients with ON. Multifocal VEP is more sensitive in detecting demyelinating ON and pattern VEP followed by contrast and Humphrey visual field is useful to identify fellow eye abnormality [2,8,10,32,33].
According to the ONTT, patient with typical ON does not require laboratory studies and lumbar puncture (LP) while in atypical ON careful examination is required to establish the correct treatment regimen specifically in children, bilateral cases or when systemic or infectious diseases are in doubt [2,4,8,9].
Systemic examination including CSF analysis consist of determination of total protein, albumin, IgG, IgA, IgM, glucose, lactate, cell count, microbiological/virological analysis and oligoclonal bands. Oligoclonal banding of proteins in CSF is a valuable predictor of the risk of MS. Blood culture and serological tests must be done to rule out infective and inflammatory cases such as SLE, syphilis and sarcoidosis (biopsy of accessible tissue if applicable). Chest X-ray and the Mantoux test is done in patients suspected to tuberculosis before starting steroid treatment [4,9,10,18,31].
TREATMENT
The goals of many of the existing and emerging treatments including steroid and immuno-modulatory therapy are reduction in the number and severity of attacks, and prevention of axonal loss and subsequent disability in both ON and MS [8].
Recovery of visual functions in ON is observed spontaneously within 2-3 weeks in more than 80% of patients without treatment. Vision stabilizes over months or continues to improve up to 1 year, although long-term defects in visual functions is possible [2,5,9,31,32].
According to the Optic Neuritis Treatment Trial (ONTT) protocol, each patient was randomly assigned to receive one of the following three regimens within 8 days after the onset of symptoms: (1) oral prednisolone, 1 mg/kg per day for 14 days (prednisone group); (2) intravenous methylprednisolone (IVMP) sodium succinate, 250 mg every 6 hours for 3 days, followed by oral prednisone, 1 mg/kg per day for 11 days (intravenous group); or (3) oral placebo for 14 days (placebo group). Regimens for oral and intravenous prednisolone groups were followed by a short tapering off with oral dosages consisting of 20 mg of prednisolone on day 15 and 10 mg on days 16 and 18 [1,2,4,6,10,35].
ONTT and other studies showed that the high-dose intravenous corticosteroids were effective in improving short-term visual recovery particularly for visual fields and contrast sensitivity as compared to oral prednisolone and placebo, but the difference in the rate of recovery subsided within 1 month and there was no statistically significant benefit in long-term (1year) outcome among the three groups. At 6 months, there was still a small statistically significant benefit for the intravenous regimen in contrast sensitivity, visual field, and color vision but not in visual acuity [1-4,6,9,10,31,32,35].
Based on the ONTT findings, intravenous steroids treatment is recommended when 3 or more signal abnormalities are present on MRI and reduces risk of developing MS by 2 years. It is also recommended in patients in whom there is a need for faster recovery of visual deficits (i.e., uniocular patients, employment demands, bilateral involvement and patients desiring intervention) [2,4,10,32].
- Visual Acuity: In the ONTT visual acuity at 1 year was 20/40 or better in 95%, 94%, 91% in placebo, IVMP and oral prednisone groups respectively [1,4].
79% and 93% of patients began to show enhancement of vision within 3 and 5 weeks of onset, 93% (69%) showed VA of >20/40 (>20/20) in the affected eye at 1 year and at 15 years follow up 72% (>92%) showed VA of ≥ 20/20 (20/40) in affected eye and 66% (1%) showed VA of ≥20/20 (<20/200) in both eyes respectively [2,4].
The risk ratio of normal visual acuity in the intravenous group compared to placebo was 1.08 (95% CI 0.89 to 1.31) at one month, 1.06 (95% CI 0.89 to 1.27) at six months and 1.06 (95% CI 0.92 to 1.22) at one year. The relative risk of normal visual acuity in the oral group compared to placebo was 0.95 (95% CI = 0.77 to 1.18) at one month, 0.93 (95% CI = 0.76 to 1.13) at 6 months and 0.76 (95% CI = 0.63 to 0.92) at 1 year [3].
While good visual function recovery is reported in most patients, approximately 5% to 10% of patients fail to recover fully. Subtle symptoms such as blurred, washed out vision may persist even in patients with VA of ≥20/20 [2,4,10].
- Contrast sensitivity: The risk ratio of normal contrast sensitivity in the intravenous group compared to placebo was 1.06 (95% CI 0.95 to 1.17) at one month, 1.10 (95% CI 0.92 to 1.32) at six months, and 0.99 (95% CI 0.93 to 1.06) at one year. The relative risk of normal contrast sensitivity in the oral group compared to placebo was 1.00 (95% CI = 0.90 to 1.12) at one month, 1.02 (95% CI = 0.83 to 1.25) at six months and 0.93 (95%CI = 0.86 to 1.00) at one year [3].
- Visual field: The pooled risk ratio of normal visual field for the intravenous group compared to placebo was 1.43 (95%CI 1.12 to 1.84) at one month, 1.08 (95% CI 0.96 to 1.22) at six months, and 1.02 (95%CI 0.86 to 1.20) at one year. The relative risk of normal visual field for oral group compared to placebo was 1.16 (95% CI = 0.88 to 1.51) at one month, 1.00 (95%CI = 0.87 to 1.14) at six months and 0.94 (95% CI = 0.79 to 1.12) at one year [3].
According to ONTT, RAPD may disappear when visual recovery is full [2].
Raz et al. reported that visual form processing has a quick (4months after onset of ON) and full recovery as compared to visual motion [20].
Studies have shown IV dexamethasone (200 mg once daily for three days) has equal effectivity as IVMP (as recommended by ONTT) with fewer side effects, easier administration and lower cost [4,31,32].
ONTT reported mild side effects of steroids such as depression, acute pancreatitis, weight gain, sleep disturbances, mild mood changes, stomach upset, facial flushing, as well as serious side effects which were rare and only happened in the IVMP group. Avascular necrosis of the hip or other joints is a serious complication that rarely occurs due to a brief course of corticosteroids. Other studies reported hyperglycemia, constipation, diarrhea, acneiform eruption, hyperlipidemia, headache and fever [3,8,35].
Intravenous immunoglobulin (IVIG) treatment and plasma exchange show contradictory results in improvement of visual function and reduction in rate of conversion to MS. Uhthoff’s symptoms are completely reversible and not damaging to vision. They can be eased by staying indoors on hot and humid days and drinking abundant cool fluids [2,4,8,9].
Immunnomodulatory Therapy
Since there is evidence of early axonal damage in acute demyelinating ON, long term treatment with disease modifying drugs (DMDs) such as interferon β-1a (Avonex®), interferon β-1b (Betaseron®) and glatirimer acetate (Copaxone®) should be considered in patients at high risk of developing MS as prophylaxis confronting permanent neurological impairment. DMDs are playing the role in increasing the time to start the second episode, and the incidence of subsequent MS relapses and demyelinating lesions. The different mechanisms suggested include reduced antigen presentation, inhibition of pro-inhibitory cytokines and autoreactive T cells, induction of immunosuppressive cytokines and reduced migration of cells in the CNS [2,4,5,8,9].
Before assigning a patient to treatment with interferon, it is important to consider that over 40% of patients with ON and an abnormal MRI scan will not develop CDMS at 10years. In addition, to prevent one relapse patients need treatment for approximately 6 years and finally the long-term visual prognosis is desirable even if MS progresses. In the USA patients are recommended to be referred to a neurologist and have a discussion about DMDs therapy while in the UK the policy remains that interferon-β or Copaxone® are not started unless a second clinical attack occur within 2 years after onset of ON [2,8].
CHAMPS (Controlled High-Risk Subjects Avonex® Multiple Sclerosis Prevention Study) was a randomized, double blind assessment including 383 patients with an early, acute mono symptomatic demyelinating event and at least 2 silent T2 lesions on brain MRI. Within 27 days after the onset of symptoms, patients were randomly assigned to one of two treatment regimen: Initial treatment with IVMP and an oral taper subsequent to weekly intramuscular injections of 30 microgram of interferon β-1a (Avonex®). Same initial treatment followed by weekly injections of a placebo. The treatment group experienced a reduction in the progression rate of CDMS compared to the placebo group (35% vs 50 %) over 3 years of follow-up as well as valuable effects on all MRI parameters, including decrease in T2 lesion development and volume, and gadolinium-enhancing lesions. Patients treated quickly with interferon β-1a after the first attack, had a lesser chance of developing a second episode within 10 years follow up compared to those who had delayed treatment (after about 30 months). The most common side effects of Avonex® were flu-like symptoms including myalgia, fever, fatigue, headache, chills, nausea, vomiting, pain and asthenia [1,4,10].
In an Early Treatment of MS study (ETOMS), 308 patients with initial clinical demyelinating events (98 of whom had acute optic neuritis) randomly received either 22 microgram weekly of interferon β-1a (Rebif®) subcutaneously or placebo. Treatment was started within three months of symptom onset; 39% of patients had two or more CNS lesions at presentation. 70% of patients received corticosteroids (variable dose and route of administration) before interferon β-1a. MS was found to be less developed in patients who received interferon β-1a compared to the placebo group (34% vs 45%) with significantly fewer new lesion formations on T2-weighted MRI at two years follow-up [10].
The most recent study that used Betaferon in Newly Emerging Multiple Sclerosis for Initial Treatment (BENEFIT), reported reduction in risk of MS by 50% within 24 months in patients who had a single neurologic event and at least 2 clinically silent MRI lesions after receiving standard dose of Betaseron® [4,10].
RISK OF RECURRENCE OF ON
Optic neuritis can occur as a monophasic or recurrent disease, either in the same or the contralateral eye especially in patients who develop MS thereafter. The ONTT reported 28% and 35% of patients recurred ON within 5 to 10 years respectively [2,14]. At the 5 year follow-up, the relapse of ON was 19% for the affected eye, 17% for the fellow eye and 30% for either eye [1].
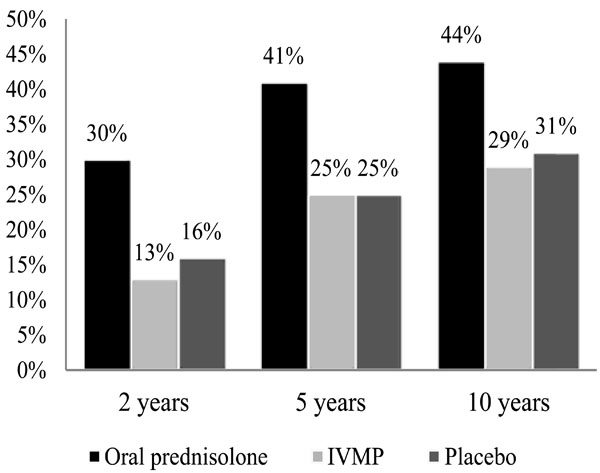
Treatment with oral prednisone alone in standard doses increased the relapse rate of ON and therefore is not recommended in acute typical ON (Table 6). Higher doses of oral corticosteroids have shown the same recurrence rate compared to placebo [1,3,6,8,10,32,35].
CONVERSION FROM ON TO M.S
The presence of demyelinating white matter lesions in brain MRI scans, 3 mm or larger in diameter, ovoid, located in periventricular areas of the white matter and radiating toward the ventricular spaces has been identified as the strongest predictor for the development of MS [2,4,5,10].
In addition to absence of MRI findings, being male, having papillitis, VA of no light perception, absence of pain, retinal exudates and peripapillary haemorrhages are related to lower risk of developing MS. The risk of MS following demyelinating ON is much lesser in children than adults and estimated to be 13% at 10 years, 19% at 20 years and 26% after 40 years [2,4].
According to ONTT, treatment with IVMP followed by oral prednisone resulted in lower rates of developing MS in the first 2 years, but this effect was not continuous after year 3. It showed 16% risk of MS development at 5 years with normal brain MRI findings, 37% with 1–2 lesions and 51% with ≥3 lesions. The only statistically significant difference at 10 years was between no lesions (22% risk) and one or more lesions (56% risk), which had increased to 25% and 75% respectively at 15 years [1,2,4,5,8-10,34,35].
CONCLUSION
The most common cause of acute optic neuropathy in young women is ON which can be either typical, in which diagnosis is carried out clinically, or atypical which necessitates complete laboratory and neurological examinations in accordance with ONTT recommendations to discover the source of inflammation. Several conditions present with identical symptoms as ON and they should be considered for appropriate treatment. The majority of patients with ON recover visual function spontaneously. However, IVMP can accelerate the rate of recovery. Most patients may experience some long-term visual defects even after receiving treatment and achieving VA of 6/6. The less expensive IV dexametasone treatment can be used as an alternative to IVMP. If any demyelinating lesions are present in MRI, the patient should consult a neurologist regarding treatment with DMDs as prophylaxis to decrease the risk of developing MS with close monitoring.
ACKNOWLEDGEMENT
The authors wish to express their gratitude to Dr. Changiz Geula, Professor of Neuroscience, Director, Laboratory for Cognitive and Molecular Morphometry Cognitive Neurology and Alzheimer's Disease Center, Northwestern University, Feinberg School of Medicine, who was abundantly offered invaluable guidance which brought this work to fruition.
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflicts of interest.

