All published articles of this journal are available on ScienceDirect.

Autoimmune Retinopathy in Systemic Lupus Erythematosus: Histopathologic Features
Authors Info & Affiliations
Abstract
The ocular pathology of autoimmune retinopathy is demonstrated in a 62-year-old female patient with systemic lupus erythematosus (SLE) who presented with typical clinical autoimmune retinopathy. Macroscopically, there were multiple depigmented lesions in the peripheral retina and choroid and scattered pigmentary bone-spickling at the equator and periphery. Microscopically, there were generalized loss of photoreceptors and thinning of the outer plexiform layer. Many peripheral retinal vessels were sclerotic and occluded, some surrounded by pigment granules and RPE cells. Cobblestone degeneration was prominent in the periphery. Macrophages were seen in the retina, particularly in areas of photoreceptor degeneration. Rare, scattered T- lymphocytes were present in the retina and choroid, while B-cells were notably absent. The optic nerve showed loss of axons and thickened septae. Serum autoantibodies against normal retinal nuclei were detected. These pathological changes represent both known SLE-associated ocular complications as well as possible features of autoimmune retinopathy secondary to SLE.
INTRODUCTION
Systemic lupus erythematosus (SLE) is an autoimmune disorder characterized by abnormal deposition of circulatory immune complexes in blood vessel walls. The deposition causes inflammation through activation of the complement system, resulting in tissue damage and disease mani-festations [1]. The disease affects mainly the joints, skin, kidneys and cardiopulmonary system. However, other organs, such as the liver, spleen, brain and eyes may be involved as well [2]. Autoantibodies produced by the cooperation of B and CD4 T-cells are a central feature of SLE. Ocular complications of SLE are well documented, including keratoconjunctivitis sicca, retinal vascular disease with vasculitis and vascular occlusion, choriodopathy, and optic neuritis [1, 3].
Autoimmune retinopathy (AIR) is an immunological process whereby retinal autoantibodies attack the retina tissue, resulting in retinal degeneration and eventual blindness [4]. Three main subtypes of AIR have been identi-fied: cancer-associated retinopathy (CAR), melanoma-associated retinopathy (MAR), and, less commonly, non-neoplastic autoimmune retinopathy (npAIR). In addition, AIR is hypothesized to play a role in retinitis pigmentosa, ocular trauma, birdshot retinopathy, acute zonal occult outer retinopathy (AZOOR), and multiple evanescent white dot syndrome (MEWDS) [4]. Reports of the ocular histopatho-logic changes in AIR are limited. Herein we describe the ocular histopathology of a patient with AIR secondary to SLE.
CASE
The study was approved by the National Eye Institute Institutional Review Board for human subjects, and informed consent was obtained from the patient. The case involves a 62-year-old white female with SLE who died of progressive multifocal leukoencephalopathy caused by JC virus within the central nervous system. She was on hydroxychloroquine (Plaquenil) for SLE for 8 years. Laboratory analysis revealed the following positive serum antibodies: antinuclear antibody (ANA), Smith (Sm, a nuclear ribonucleoprotein that is specific to SLE), ribonucleoprotein (RNP, nuclear or cytoplasmic), as well as positive lupus anticoagulant. The serum tested negative for β-2-glycoprotein-1 (β-2-GP1, the main affecter of cardiolipin binding) and anticardiolipin antibody (ACA). Two weeks prior to her death, the patient’s behavior suggested that she had experienced visual decline. Ocular testing revealed a best-corrected visual acuity of 20/50 OD and 20/100 OS, color vision by Ishihara plates of 13/16 OD and 11/16 OS, and contrast sensitivity in log units of 1.35 OD and 1.05 OS. Intraocular pressure was 17mmHg OD and 14mmHg OS. Posterior subcapsular cataracts were noted bilaterally. Fundus examination revealed a blunted foveal light reflex, peripheral “punched out” retinal lesions, and bone spickling bilaterally (Fig. 1). The patient was too ill to perform electrophysiological testing in clinic. A clinical diagnosis of AIR secondary to SLE was suspected. The patient’s eyes were obtained for autopsy.

Fundus photographs. The color funduscopy of the right eye (R) and left eye (L) show blunted foveal light reflex, peripheral “punched out” lesions (white arrows) and bone spickling lesions (black arrows) bilaterally.
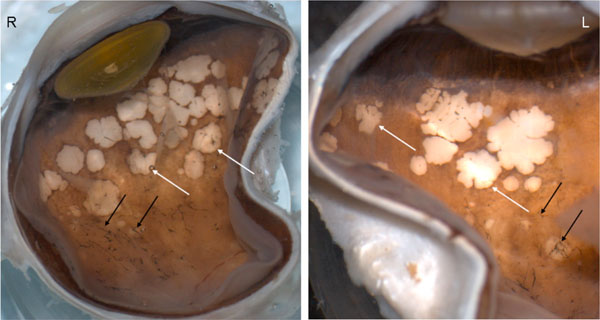
Macroscopic photograph. There are multiple pigmentary bone-spickling lesions (black arrows) in the equator and peripheral retina of both eyes (R, Right eye; L, Left eye). Multiple medium- to large-sized focal depigmentary cobblestone lesions (white arrows) are also observed in the periphery.
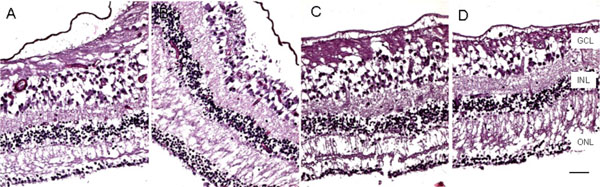
Microscopic macula. The retina is markedly atrophic characterized by generalized loss of photoreceptors and thinning of the outer plexiform layer except the temporal maculae from fovea to equator of the right eye (A, nasal; B, temporal), but worse in the left eye, even at the maculae (C, nasal; D, temporal). (GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; scale bar indicates 50 µm).
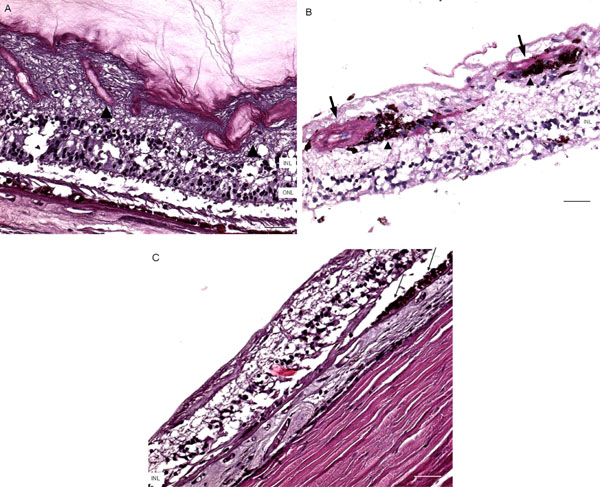
Microscopic retinas. A, some small retinal vessels are sclerotic (arrowheads) in the right eye. B, some occluded vessels (arrows) are surrounded by pigment granules and a few migrated RPE cells (arrowheads) in the right eye. C, The left eye shows an absence of choriocapillaris, RPE, photoreceptors and the outer nuclear layer, while the remaining inner nuclear layer was adherent to an irregular Bruch’s membrane. Hypertrophic RPE (arrows) was present at the margin of these lesions. (GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; scale bar indicates 50 µm).
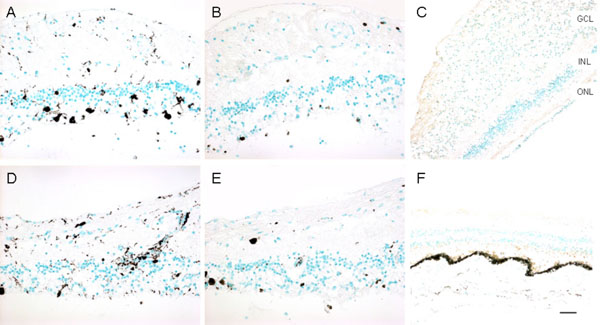
Imunoreactivities against CD68, CD3, and CD20 markers in the retinas. A, CD68, right eye; B, CD3, right eye; C, CD20, right eye; D, CD68, left eye; E, CD3, left eye; F, CD20, left eye. CD68 positive cells are scattered in the retina, while most are where photoreceptor are lost. Rare, scattered CD3 positive cells are present in the retina. CD20 staining is negative in both eyes (Green = methyl green counterstain; brown = positive immunostain). (GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; Scale bar indicates 50 µm).
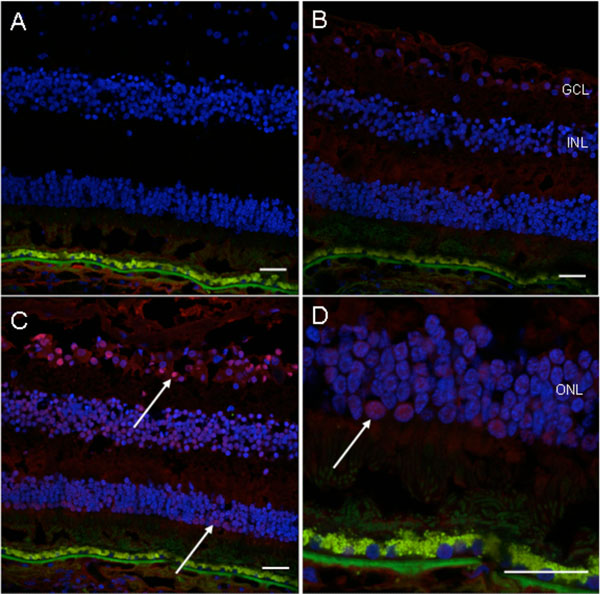
Immunofluorescence detection of autoantibody in the patient’s serum on the normal retinal sections. A, no serum incubation (control); B, normal human serum incubation (1: 100, control); C, patient’s serum incubation (1: 100); D, higher magnification photo of C. There is positive fluorescence in the ganglion cell layer, inner nuclear layer and outer nuclear layer (white arrows) of the normal human retinal section incubated with the patient’s serum (Blue fluorescence = nuclear, DAPI staining; green fluorescence = autofluorescence; red fluorescence = positive staining). (GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; scale bar indicates 50 µm).
Routine histopathology and immunohistochemistry were performed on both globes. Immunohistochemistry was conducted using the avidin-biotin-complex immunoperoxidase tech-nique with the Betazoid DAB Chromogen Kit (Biocare medical, CA, USA). Primary antibodies included CD68 (macrophage and microglia), CD3 (T-lymphocyte), and CD20 (B-lymphocyte) (Dako North America, Inc., Carpinteria, CA, USA). Secondary antibody was biotinylated goat anti-mouse IgG (1: 200; Vector Laboratories, CA, USA). Confocal immunofluorescence was performed to detect autoantibodies in the patient’s serum, as described previously [5]. The technique in brief was as follows: normal human retinal sections were incubated with patient’s serum (1: 100), normal human serum (1: 100), or no serum, and then incubated with fluorescent secondary antibody (Alexa Fluor® 568 goat anti-human IgG (H+L), Invitrogen, USA). Immunoreactivity of each slide was assessed under a confocal microscope with the same confocal settings (Leica, Wetzlar, Germany). The images have been normalized to background fluorescence prior to visual comparisons.
Macroscopically, there were multiple pigmentary bone-spickling lesions in the equator and peripheral retina of both eyes. Multiple medium- to large-sized focal depigmentary cobblestone lesions were also observed in the periphery (Fig. 2). The optic nerve heads were pale. Microscopically, the lenses had posterior epithelial migration and posterior subcapsular globulins. The outer layers of the retina were diffusely atrophic and characterized by generalized loss of photoreceptors and thinning of the outer plexiform layer, sparing the temporal macula from the fovea to the equator of the right eye, but involving the entire macula of the left eye (Fig. 3). In the periphery, many retinal vessels were sclerotic and occluded in both eyes (Fig. 4). Some of the occluded vessels were surrounded by pigment granules and a few migrated RPE cells. Typical cobblestone degeneration was found in the temporal and peripheral retina of both eyes, characterized by an absence of choriocapillaris, RPE, photoreceptors and the outer nuclear layer, while the remaining inner nuclear layer was adherent to an irregular Bruch’s membrane. Hypertrophic RPE was present at the margin of these lesions. Macrophages and microglia were prominent in the retina where photoreceptors had been lost (Fig. 5A, d), as well as in the choroid. Rare T-lymphocytes were scattered throughout the retina and choroid in both eyes (Fig. 5B, E). Interestingly, there was no B-lymphocytic infiltration (Fig. 5C, F). The optic nerves demonstrated loss of axons and thickened septae, as well as mild to moderate macrophage infiltration and microglial accumulation. Positive immunoreactivity of patient’s serum was detected in the ganglion cell layer, as well as both inner and outer nuclear layers; in contrast, immunoreactivity was negative for control normal serum, which indicated the presence of serum autoantibodies against retinal cellular nuclei including the photoreceptor (Fig. 6).
DISCUSSION
SLE is a chronic inflammatory disease of unknown origin with the ability to affect many organs. In a study of 550 patients with SLE, Stafford-Brady and colleagues observed retinopathy in 41 patients (7.1%) [6], while a study of children with SLE reported ocular involvement in 35% of patients [7]. In the case of our patient, histopathologic examination of the peripheral retina revealed many small occluded vessels, some appearing similar to changes seen in chronic retinal vasculitis [8]. The vascular occlusive findings and cobblestone degeneration are likely the consequence of SLE vasculopathy and lupus anticoagulant thrombosis.
Histopathology of our patient’s eyes demonstrates diffuse loss of photoreceptors, including areas exhibiting normal retinal and choroidal vasculature. This finding suggests that the retinal abnormalities may not result from the common ocular complication of vascular inflammation and occlusion. Since SLE is associated with loss of B-cell tolerance, B-cell dysregulation, and high-affinity autoantibody production [9], the clinical and pathological presentation of our patient suggests that AIR could result from severe SLE. Although her serum demonstrated immunoreactivity not only against photoreceptor, but also other retinal cell nuclei, which may relate to elevated serum ANA; we could not rule out the presence of specific autoantibody targeting the photoreceptor in this case.
Antibodies against retinal antigens have been reported in patients with cancer, multiple sclerosis, and presumed in SLE, although AIR per se has not been documented in SLE patients [10-12]. The autoantibody response in SLE is both diverse and exuberant, targeting a variety of nucleoprotein particles, some containing DNA (nucleosomes), and others RNA. The latter include small nuclear ribonucleoproteins (RNPs), cytoplasmic RNPs, and ribosomes. Antibodies are also directed against negatively charged phospholipids. A striking feature of the different autoantigens in lupus is their lack of restriction to a single subcellular location. Since the retina develops embryologically as an extension of the neural tube, it shares with the neural tissue several cell membranes- and cytoplasm-associated antigens. It would seem possible, therefore, that patients with SLE could produce auto-antibodies targeting neural-specific antigens, inducing neural tissue damage such as that seen in the retina of our patient [12-14]. We would therefore suggest our patient’s condition as npAIR.
Our patient was treated with Plaquenil, which has the potential to induce a toxic retinopathy [15] in a distinctive macular pattern not seen in our patient. She was also diagnosed with progressive multifocal leukoencephalopathy, a demyelinating brain disease caused by the JC papovavirus. This infection usually progresses rapidly, causing a variety of visual and neurologic complications. However, no association between progressive multifocal leukoencephalo-pathy and AIR has been published in the literature.
The clinical spectrum of AIR is heterogeneous and diverse. The fundus may appear normal or demonstrate characteristics of retinal degeneration such as attenuated retinal vessels, waxy disc pallor, and RPE mottling or atrophy [4]. Signs of inflammatory activity are classically absent [16]. The primary features that suggest an auto-immune component are the negative waveform on ERG [17] and visual field loss or scotomata [13]. The diagnosis is based in part on the detection of serum antiretinal antibodies in the setting of clinical and electrophysiological evidence of retinal degeneration [14, 18, 19]. If serum antibodies are identified against retinal tissue, and clinical symptoms and findings are compatible with retinal degeneration; a tentative diagnosis of AIR can be made. The hallmark of AIR is the subacute onset of symptoms, rapid progression of vision loss, and an abnormal ERG in a patient with an appropriate medical history.
AIR is associated with circulating antibodies directed against retinal proteins/antigens; however, the precise pathogenesis of AIR is still unknown. In AIR, the inflammation is usually minimal. In CAR, the targeted antigen is recoverin, a calcium-binding protein found in both rods and cones. In vitro experiments have shown that antibodies against recoverin may cause blockage of ion channels and cellular depolarization, with eventual photoreceptor cell death. When anti-recoverin antibodies enter photoreceptor cells, apoptosis occurs without severe inflammation [16, 20].
The findings of mild ocular inflammatory cells on histopathology in our case are intriguing although not unexpected. Macrophages and microglia in the areas of neuronal cell loss (both photoreceptor and optic nerve) presumably function to phagocytose the dead neurons and to repair tissue damage. Interestingly, scattered T-, but not B-lymphocytes reflect the ocular pathology at the late stage of AIR. It is possible that autoantibodies were produced by plasma cells and B-lymphocytes in the lymph node, circulated into the eye and attacked the photoreceptors without direct ocular infiltration of B-lymphocytes or plasma cells. Therefore, general immunosuppresion, in addition to therapy targeting B-lymphocytes, such as rituximab, should be considered in the management of AIR.
CONCLUSION
This report highlights the ocular histopathological features of wide-spread loss of photoreceptor and mild mac-rophage infiltration and microglia accumulation resulting from AIR. These findings may help improve our under-standing of the pathogenesis of this disease.
ACKNOWLEDGEMENTS
The Intramural Research Program of the National Eye Institute, NIH provided the funding support of the study.

