All published articles of this journal are available on ScienceDirect.

Controlled Release of Bevacizumab Through Nanospheres for Extended Treatment of Age-Related Macular Degeneration
Authors Info & Affiliations
Abstract
Bevacizumab (Avastin®) has been used by ophthalmologists in many countries as an off-label drug for the treatment of wet age-related macular degeneration (AMD). Due to its short half-life necessitating frequent intravitreal injection, a method for sustained delivery is in need. We demonstrated that bevacizumab could be released in a sustained fashion over 90 days from nano- and microspheres fabricated from poly(DL-lactide-co-glycolide) and poly(ethylene glycol)-b-poly(D,L-lactic acid), respectively. The drug release rate could be adjusted by alteration of the drug/polymer ratio. The use of such nano- and microspheres as bevacizumab delivery vehicles may improve the treatment of wet AMD.
1. INTRODUCTION
In the past few years, intravitreal injection of an anti-cancer drug, bevacizumab (Avastin®, Genetech) [1] has been found to be a very effective treatment for the wet form of age-related macular degeneration (AMD) [2], proliferative diabetic retinopathy [3] and choroidal neovascularization [4]. Short-term results suggested that intravitreal bevacizumab (1.25mg) was well tolerated and associated with improvement in visual acuity, decreased retinal thickness and reduction in angiographic leakage in most patients [5]. Bevacizumab is thought to be as safe as ranibizumab (Lucentis®, Genetech), a prescription medication approved for the treatment of wet AMD [6]. It has been increasingly used in many countries as an off-label treatment for AMD and is evolving into the standard of care for wet AMD. However, to achieve and maintain the improvement in vision, repetitive injection and follow-up visits are required. Intravitreal injection is a currently accepted therapy for many posterior segment ocular disorders. However, when repeated injections are required, there is a high risk of complications such as endophthalmitis [7], as well as repetitive pain, apprehension and distress associated with inserting needles into eyes. Moreover, the intravitreal half-life of 1.25mg injected bevacizumab is approximately only 3 days [8]. Therefore an effective drug delivery method needs to be developed to render bevacizumab delivery less invasive and long-lasting.
Recently, nanospheres, microspheres and liposomes have been extensively studied as drug carriers in the pharmaceutical and medical fields [9-14]. They have shown promise as drug delivery systems due to their controlled and sustained release properties, subcellular size and biocompatibility with tissue and cells. Very recently, studies have shown the beneficial effects of liposomes in prolonging the residency of bevacizumab in the vitreous in animal model during 42 days [15]. Hao et al. [16] encapsulated bevacizumab within Poly(DL-lactide-co-glycolide) (PLGA) nanoparticles with an entrapment rate of 45%. The release of the entrapped bevacizumab from the nanoparticles could last 4 weeks. Pan et al. [17] also created long-lasting formulations (8 weeks) of bevacizumab through poly(ethylene glycol) (PEG) conjugation or encapsulation within PLGA nanoparticles. In this study, we encapsulated bevacizumab within particles of PLGA and poly(ethylene glycol)-b-poly(D,L-lactic acid) (PEG-b-PLA), and studied the profiles of drug release from these different polymers.
2. MATERIALS AND METHODS
2.1. Materials
Bevacizumab (25mg/ml, Avastin®, Genentech Inc) was purchased from the Ottawa Hospital Pharmacy (Ottawa, Canada). Poly (DL-lactide-co-glycolide) (50:50) (PLGA, Mw=17,000-22,000Da), PEG(1000)-b-PLA(5000) (PEGLA 15) and PEG(5000)-b-PLA(5000) (PEGLA55) were purchased from PolySciences Inc. (PA, USA).
2.2. Preparation of Bevacizumab-Loaded PLGA Nanoparticles (PLGA-A)
305.2mg PLGA was dissolved in 5ml of dichloro-methane. To this solution, 0.2ml of bevacizumab (25mg/ml) was added. The resulting mixture was sonicated for 30 seconds using a probe sonicator (Model VC50, Sonics & Materials Inc, Danbury, CT, USA). Then, the mixture was poured into 40ml of polyvinylpyrrolidone solution (2.5% w/v) and was sonicated for another 30 seconds. After evaporation of dichloromethane through stirring the mixture overnight in fume hood, nanoparticles were collected through centrifugation and washed with 25ml x 2 MiliQ water. Finally, nanoparticles were freeze-dried overnight, in 74% yield. Bevacizumab loading efficiency (LE) is 95.4% as determined by UV spectrometry based on the following equation:
LE= C/Co x 100%
where, C is the quantity of encapsulated bevacizumab - calculated by subtracting the quantity of bevacizumab in the supernatant and the washing solutions from the initial feeding quantity of bevacizumab, Co.
All other bevacizumab-loaded particles were prepared as described above. For size comparison, bevacizumab-loaded PLGA particles were also prepared by homogenization of the corresponding mixture using a homogenizer (ULTRA-TURRAX, IKA T10 Basic, IKA Works Inc, USA).
2.3. Bevacizumab Release Study
Between 30-50mg of bevacizumab-loaded particles were placed into centrifuge tubes, followed by addition of 1ml of 10mM phosphate buffered saline (PBS). The tubes were capped and maintained in a 37oC incubator. At certain intervals, the tubes were centrifuged and 0.5ml of the supernatant was removed followed by addition of an equal volume of fresh PBS. The concentration of bevacizumab in the supernatant was determined using a UV spectrophotometer (Beckman 640B) at 270nm against a calibration curve.
3. RESULTS
3.1. Bevacizumab-Loaded Nano- and Microparticles
For equivalent bevacizumab/PLGA ratios (1.6%), the particle size range obtained was dependent upon the method of preparation. PLGA spheres were both obtained but the particle sizes were heterogenous both when a probe sonicator was used (Fig. 1), and when a homogenizer was used (Fig. 2). Sonication resulted in particles with diameters mainly within the 0.2 to 1.0µm range. Particles prepared using a homogenizer, however, were more polydispersed, with the majority of particle diameters ranging from 0.5 to 3µm. When the loading of the drug was increased to bevacizumab/PLGA = 13%, the particles no longer showed shape uniformity, although some spheres were still present (Fig. 3). The particles fabricated from PEGLA15 appeared spherical and porous (Fig. 4), with the diameters of the particles ranging from 2 to 10µm. When PEGLA55 polymer was used, the diameter of particles was under 1µm, but the particles were no longer spherical (Fig. 5).

SEM image of bevacizumab-loaded PLGA nanoparticles (bevacizumab/PLGA= 1.6 %) prepared through sonication using probe sonicator.

SEM image of bevacizumab-loaded PLGA particles (bevacizumab/PLGA= 1.6 %) prepared through homogenization using homogenizer.

SEM image of bevacizumab-loaded PLGA nanoparticles (bevacizumab/PLGA= 13 %) prepared through sonication using probe sonicator.

SEM image of bevacizumab-loaded PEGLA15 particles (bevacizumab /PEGLA15=1.9%) prepared through sonication using probe sonicator.

SEM image of bevacizumab-loaded PEGLA55 particles (bevacizumab/PEGLA55= 2.0 %) prepared through sonication using probe sonicator.
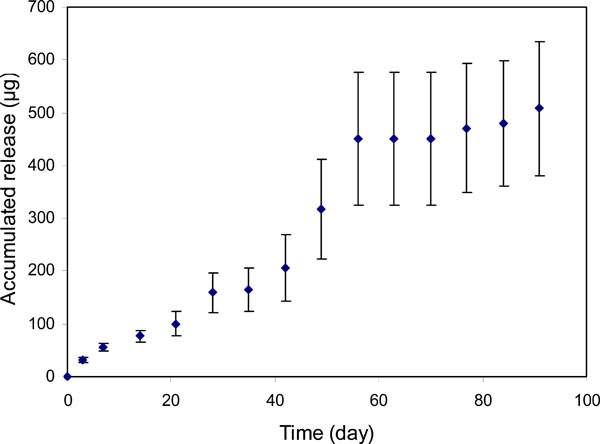
Profile of bevacizumab release from PLGA nanoparticles (bevacizumab/PLGA=1.6%).
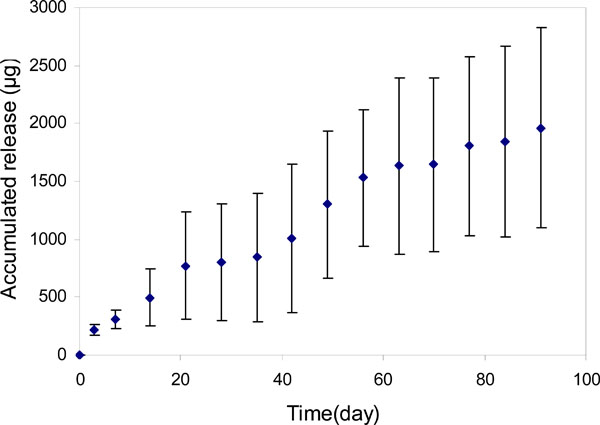
Profile of bevacizumab release from PLGA nanoparticles (bevacizumab/PLGA=13.0%).
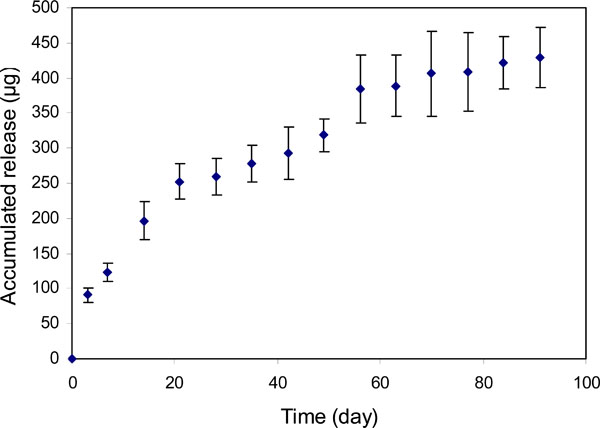
Profile of bevacizumab release from PEGLA15 particles (bevacizumab/PLGA=1.9%).
Within our experimental range, the drug encapsulation (loading) efficiency was above 90% (Table 1) when PLGA and PEGLA15 polymers were used. The yield of the particles was greater than 70% with these two polymers. However, when PEGLA55 was used, the yield dropped to 24%.
Drug Encapsulation (Load) Efficiency
| Particle Types | Yield, % | Load Efficiency, % | Drug/Polymer, % | Particle Size, µm |
|---|---|---|---|---|
| PLGA-A* | 74 | 95.4 | 1.6 | 0.2-1 |
| PLGA-HiA | 80 | 96.4 | 12.9 | < 1 |
| PEGLA15-A | 99 | 93.6 | 1.9 | 2-10 |
| PEGLA55-A | 24 | NA | 2.0 | < 1 |
* A represents bevacizumab.
3.2. Bevacizumab Release Profiles
For the particles with an initial bevacizumab/ PLGA=1.6% loading ratio (PLGA-A, Table 1), approximately 0.5mg of bevacizumab was relatively steadily released over 55 days (Fig. 6). Beyond this time until day 91, only an insignificant amount of release was detected. However, when the bevacizumab/PLGA ratio was increased to 13% (PLGA-HiA, Table 1), a steady release of 1.9mg of bevacizumab was observed throughout the experimental period of 91 days. There was a plateau in the release profile around 30 days for both of the release curves (Figs. 6, 7). Over the 91-day release period, we calculated that approximately 47% and 64% bevacizumab were released from particles with 1.6% and 13% drug loading respectively. Bevacizumab release from the PEGLA15 particles showed a similar plateau in the release profile (Fig. 8) as those in Figs. (6) and (7), but starting at about day 20. Approximately 62% of encapsulated bevacizumab was released from this system during the 91-day period.
4. DISCUSSION
PLGA is an FDA-approved biodegradable polymer, and has been extensively studied for its biocompatibility, toxicology and degradation kinetics [18, 19]. It has been used clinically as a suture material since the 1970s [20] and it has been recently used as scaffolds in tissue engineering [21,22]. More importantly, the degradation rate of PLGA is in the range of months to years [23]. This is dependent upon the ratio of lactide to glycolide units in the polymer [24,25], which is favorable for regulating the release rate of encapsulated drugs. PLGA nanospheres or microspheres have been studied as vehicles for intravitreal delivery of various types of drugs such as dexamethasone acetate [26], ganciclovir [27], 5-fluorouracil [28] and glial cell line-derived neurotrophic factor [29], as well as for transscleral delivery of Macugen® (pegaptanib sodium), the first anti-VEGF inhibitor that was approved for wet AMD treatment [30]. These in vivo studies for treatment of various ocular diseases have reported no sign of retinal toxicity nor significant inflammatory responses for periods of up to 2 months [28,31]. Therefore, in this work we selected PLGA for encapsulating bevacizumab to achieve sustained release of this drug.
We noted that with a higher loading of bevacizumab, the PLGA particles lost uniformity, this was most likely due to partial phase separation of PLGA polymer and bevacizumab. PEG-b-PLA has two polymer blocks. The PEG block is water-soluble and has anti-fouling properties. As a result, it is believed that the particles prepared from this polymer are long-circulating within the in vivo environment. Unlike PLGA, PEGLA15 is hydrophilic and retains water. Water could be removed via freeze-drying, which was why the particles from this polymer appeared porous after loss of absorbed water. The hydrophilicity of PEGLA15 also facilitated dispensing the particles compared to PLGA. The PEGLA55 polymer was even more hydrophilic than PEGLA15. This resulted in a lower yield of the particles as the majority of the polymer was dissolved in the water phase during particle preparation.
It is noted that the use of liposomes was reported for enhancing the in vivo residence of bevacizumab in animal model [15]. However, the reported drug encapsulation efficiency was only about 45%. Hao et al. [16] also reported a similar drug encapsulation efficiency using ethyl acetate and phosphate buffer as oil and aqueous phases respectively. However, in our approach, more than 90% drug encapsulation efficiency was achieved via double emulsion methods using PLGA and PEGLA15 polymers.
The plateaus in the release profiles at around 30 days for both of the release curves as shown in Figs. (6) and (7), suggested that the release of the surface absorbed bevacizumab was completed after about 30 days of incubation of the particles. After that, the PLGA began degrading and released additional drug entrapped within the polymeric core [32].
5. CONCLUSION
Our results indicated that bevacizumab can be released from nano- and microparticles of PLGA and PEGLA in a sustained fashion for over 91 days. The release rate could be adjusted by altering the drug/polymer ratio. However, bevacizumab activity still needs to be tested in vivo using animal models. Nevertheless, these results may offer improvements for the treatment of wet AMD using bevacizumab and may also improve cancer treatment using the same carriers for sustained release of the drug.
ACKNOWLEDGEMENT
Declared none.
CONFLICT OF INTEREST
Declared none.

