All published articles of this journal are available on ScienceDirect.

A Narrative Review of Morquio Syndrome: Mucopolysaccharidosis (MPS) Type IV
Authors Info & Affiliations
Abstract
Background:
Morquio Syndrome or Mucopolysaccharidosis Type IV (MPS IV) is a rare inherited metabolic disorder characterized by the deficiency of certain lysosomal enzymes involved in the breakdown of mucopolysaccharides. The deficiency results in proteoglycan accumulation, particularly keratan sulfate in various tissues in the body. Morquio Syndrome is classified into two subtypes: Morquio A and Morquio B, which are distinguished by the specific enzyme deficiency involved, with Morquio A being more prevalent. Morquio Syndrome commonly affects the eyes with 46.7% of diagnosed patients experiencing ocular symptoms related to the disorder.
Objective:
To perform a comprehensive review of the existing literature and summarize the ophthalmological manifestations of Morquio Syndrome.
Methods:
A methodical literature search was conducted, including papers with abstracts discussing ophthalmology or ocular pathology of “Mucopolysaccharidosis Type IV” or ” Morquio Syndrome.” Twelve relevant articles met the inclusion criteria and were included in the review. Seven of the articles consisted of case reports, collectively reporting on forty-one patients with Morquio Syndrome, primarily Morquio A. The outcome of the narrative review is an overview of the existing literature on ocular presentations of Morquio Syndrome and a summary of case report findings.
Results:
Forty-one different patients were identified from the included case reports, and forty patients were included as they presented with ocular pathology related to Morquio Syndrome. Corneal opacification was the most common presentation where twenty-seven patients had significant corneal opacification and seven patients had slight corneal opacification. Small, gray, spherical, dust-like opacities that dispersed among the stroma were most commonly seen in cases with corneal opacification. Other ocular presentations included decreased visual acuity, astigmatism, lens opacities, and glaucoma.
Conclusion:
Morquio syndrome, a rare genetic disorder, exhibits multiple ocular symptoms, with corneal opacification being the most common. While most research has concentrated on Morquio A, the more severe subtype, there's limited information on Morquio B, highlighting a need for more comparative studies. As the syndrome remains incurable, exploring new treatment avenues and understanding the reasons behind these ocular manifestations can significantly improve the quality of life for Morquio patients.
1. INTRODUCTION
The group of inherited metabolic disorders that is produced by deficiencies in specific lysosomal enzymes is identified as mucopolysaccharidoses. These lysosomal enzymes function to degrade certain mucopolysaccharides, and a deficiency can cause the accumulation of these type-specific mucopolysaccharides in various cells, such as the cells of the skeleton and organs. Rather than using the clinical presentation and symptoms, the several types of mucopolysaccharidosis disorders are distinguished by the specific enzyme that is deficient and the proteoglycan that accumulates within the patient’s cells as a result [1]. Both Mucopolysaccharidosis Type IV A, also known as MPS IVA or Morquio A, and Mucopolysaccharidosis Type IV B, also known as MPS IVB or Morquio B, are autosomal recessive multisystem mucopolysaccharidosis disorders that result in the build-up of the proteoglycan keratan sulfate. Proteoglycan accumulation in Morquio A syndrome is the result of a galactosamine-6-sulfatase deficiency [2], whereas an acid-β-galactosidase deficiency is the cause of proteoglycan accumulation in Morquio B syndrome [3]. As both Morquio A and B result in the build-up of proteoglycan keratan sulfate, their clinical presentations are considered to be similar. However, with Morquio A being the more severe and prevalent subtype, much of the clinical knowledge and symptom recognition on the presentation of Morquio Syndrome in general is based on the findings of Morquio A [3].
Most infants with the genetic trait for Morquio Syndrome present with no symptoms, but progression to an advanced-stage Morquio Syndrome is relatively rapid and can occur in a few years. Skeletal symptoms are typically the first to present around age three, followed by more organ and tissue symptoms presenting themselves before age five [3]. Of the mucopolysaccharidosis disorders, Morquio Syndrome has the most severe skeletal changes, including short-trunk dwarfism, scoliosis, and hypermobile joints [3, 4]. The skeletal manifestations of Morquio Syndrome can present at varying levels of severity. Factors that differentiate Morquio Syndrome from other mucopolysaccharidosis disorders include atlanto-occipital dislocation which can cause breathing paralysis, resulting in early adulthood mortality [4]. Children with Morquio Syndrome also retain their full intellectual function. Following the skeletal changes, organ- and tissue-related pathologies begin to arise, including pulmonary compromise, heart disease, hearing loss, and ocular changes [3]. More specifically, Morquio Syndrome can affect all ocular tissues (cornea, retina, optic nerve, and lens), and of the mucopolysaccharidosis disorders, the Morquio Syndrome causes the most severe opacification of the cornea and an increase in the risk of glaucoma [4].
There is currently no cure for Morquio Syndrome, but there are certain treatments that can target its symptoms [5]. Currently available treatments, such as keratoplasty, are not able to stop the accumulation of keratan sulfate, instead they may attempt to temporarily decrease the quantity of accumulation in order to improve the quality of life in the short term [4].
Most clinical reviews examine all the aspects of Morquio Syndrome and only discuss corneal opacification, but few focus on the ocular presentations despite further presentations existing. Thus, it is important to address the different ophthalmologic pathologies that arise during clinical examinations of patients diagnosed with Morquio Syndrome and their effects on quality of life. It has been found that 46.7% of diagnosed patients experience ocular symptoms related to the disorder [6] and 28% of patients experience ocular abnormalities as an initial symptom of Morquio A [5].
2. METHODS
A methodical literature search was conducted in PubMed, Ovid MEDLINE, Embase, and Cochrane using the terms “Morquio Syndrome,” “Ophthalmology,” or “Ocular” for all dates until April 15th, 2022.
Hand searching using Google scholar and forward and backward citation searching were also conducted and included in the literature base. Articles of all languages and levels of evidence were included. Papers were included if their abstracts discussed ophthalmology or ocular pathology and excluded if their content did not meet the inclusion requirements.
The study population included patients with Morquio Syndrome with ophthalmic pathology or symptoms. Case presentations were compared for context, discussing potential extraneous causes for the ocular presentation. The outcome of the review is an overview of the existing literature on ophthalmology in Morquio Syndrome and a summary of the case report findings (Table 1).
| - | Abraham et al. | Cahane et al. | Couprie et al. | Gosele et al. | Hendriksz et al. | Kasmann-Kellner et al. | Montan o et al. | Nelson et al. | Olsen et al. | Tomatsu et al., | Tomatsu et al., 2014 |
| Paper Type | |||||||||||
| Case Report |

|
|
x |
|
x |
|
x |
|
|
x | x |
| Retrospective Review | x |
|
x | x | x | x | x | x | x | x | X |
| Clinical Review | x | x | x | x |
|
x | x | x | x |
|
x |
| Survey Study | x | x | x | x | x | x |
|
x | x | x | x |
| Literature Review | x | x | x | x | x | x | x | x | x | x |
|
| Symptoms | |||||||||||
| Stromal Opacities |
|
|
x |
|
x |
|
x | x |
|
|
x |
| Corneal Clouding |
|
|
|
|
|
|
|
|
|
|
|
| Peripheral Thicken Ng of the Cornea |
|
x | x | x | x | x | x | x | x | x | x |
| Nyctalopa |
|
x | x | x |
|
|
x | x | x | x | x |
| Corneal Hypoesthesia |
|
|
x | x | x | x | x | x | x | x | x |
| Glaucoma | x |
|
x | x |
|
|
x | x | x |
|
x |
| Photophobia | x | x | x | x |
|
x | x | x | x |
|
x |
| Astigmatism | x | x |
|
x |
|
x | x | x | x | x | x |
| Punctate Cataract | x | x |
|
- |
|
|
- | x |
|
|
x |
| Myopia and Hyperopia | x | x | x | x |
|
x | x | x | x | x | x |
| Optic Nerve Atrophy | x | x | x | x |
|
|
x | x | x |
|
x |
| Shallow Orbits | x | x | x | x |
|
|
x | x | x | x | x |
| Nystagmus | x | x | x | x | x |
|
x | x | x | x | x |
| Retinopathy | x | x | x | x |
|
|
x | x | x |
|
x |
3. RESULTS
Twelve articles published from 1974 to 2014 were included in the narrative review, with eight articles sourced from PubMed, two from Embase, and one from Wiley Online Library. Nine of these articles discussed Morquio A, while two did not specify a subtype of Morquio Syndrome [7, 8], and one discussed mucopolysaccharidosis disorders in general [1].
Of the twelve articles, seven were an accumulation of case reports, totaling to forty-one different cases. Thirty-seven of the forty-one patients discussed in the case reports were diagnosed with Morquio A syndrome, and the other four patients were diagnosed with Morquio Syndrome, but their case reports did not differentiate between type A and type B. Four of the twelve articles were clinical reviews or manifestations which addressed the general diagnostics and treatment of Morquio A syndrome, with the final article being a clinical review of the different mucopolysaccharidosis disorders.
Among the forty-one patients discussed in the case reports, forty presented with some form of ocular symptoms related to Morquio Syndrome. Couprie et al. reported on twenty patients diagnosed with Morquio A, of whom twelve were females and eight were males, and their mean age was twenty-three years [9]. In the rest of the case reports spread among the six articles, sixteen patients ranged from eight months to fifteen years old at the time of the clinical evaluation. There were also two patients at thirty-six years of age, and two patients aged twenty-seven and thirty-five years at the time of the clinical evaluation [4, 7, 8, 10-12].
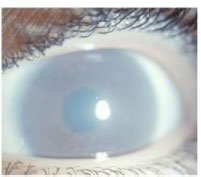
The case reports describe twenty-seven patients who had presented with significant corneal opacification, as seen in Fig. (1) [4, 7-12], and seven patients with slight corneal opacification [7, 11, 12]. In conjunction with corneal opacification, small, grayish, spherical, dust-like opacities that dispersed among the stroma were common. Some of these opacities were dispersed evenly, while others were denser toward the periphery of the stroma [4, 7, 8, 12]. Despite corneal opacification, only three patients were reported with a poor visual acuity (OD ranging from 20/100 to 25/100 and OS ranging from 16/100 to 20/100) [4, 8]; other patients had an average visual acuity of 70/100 with optical corrections for OS and OD [9].
Upon examination, twelve patients presented with astigmatism [9], and nine patients were found to have some form of lens opacities, as seen in Fig. (2) (punctate or lamellar or zonular) [9, 12].

Less commonly seen clinical signs in the case reports included two patients with high intraocular pressure, leading to glaucoma, and in certain cases, optic atrophy [4, 12], corneal thickening along the periphery, slight and moderate nyctalopia [7], hypoesthesia [7, 8], and one patient who experienced nystagmus of their left eye [4].
4. DISCUSSION
Due to enzyme deficiencies, Morquio Syndrome results in the excess accumulation of keratan sulfate within the different tissues, organs, and epitheliums [3]. This excess of keratan sulfate preferably accumulates in the bone and the stroma of the cornea, explaining why corneal opacification was the most prevalent of the ocular symptoms seen among the forty Morquio Syndrome patients. The presence of small, grayish, spherical, dust-like opacities within the lenses of multiple Morquio patients suggests that these opacities also manifest due to the accumulation of keratan sulfate. It was also suggested that these opacities are a result of a different metabolic abnormality caused by Morquio Syndrome, resulting in punctate or lamellar or zonular cataracts [12]. Various stages of corneal opacification were seen among the patients; those who only had slight corneal opacification were under the age of nine, while the two patients with the highest degree of opacification were thirty-five years old and thirty-six years old. Thus, as age progresses, the amount of keratan sulfate build-up also continues to increase, leading to a greater opacity of the cornea, and likely a decrease in visual acuity [4]. Since corneal opacification worsens progressively, the early adulthood mortality rate of Morquio Syndrome presumably impacts the low abundance of reports documenting significantly decreased visual acuity or severe ophthalmological complaints [4, 7]. In situations where corneal opacification is raising ophthalmological concerns and resulting in a decline of visual acuity, keratoplasty is a reasonable treatment option.
However, keratoplasty is only a short-term solution. It has been shown in previous clinical practices that corneal opacification overtook the corneal graft within one year post-keratoplasty, and any visual improvements began to rapidly decline. Therefore, keratoplasty is recommended as a treatment to increase quality of life for a short period, which is advantageous nonetheless given the decreased life expectancy of patients with Morquio Syndrome [4].
Unlike the stroma, Morquio Syndrome affects the trabecular meshwork more slowly and gradually increases intraocular pressure. In the case reports, the two patients diagnosed with glaucoma were in their thirties, having had ample time for their trabecular meshwork to undergo alterations and their intraocular pressure to increase. Therefore, given the worsening of ocular symptoms with increasing age, the shorter life expectancy of patients with Morquio Syndrome likely contributes to the minimal reports of Morquio-linked glaucoma [8]. Glaucoma can also lead to optic nerve atrophy which may explain the rare occurrence of atrophy seen in Morquio patients [2].
This narrative review is limited by the lack of investigations of ocular pathologies beyond corneal opacification in the literature. Thus, this review’s ability to objectively summarize the prevalence of different ocular symptoms in Morquio patients was limited.
CONCLUSION
To summarize, this review presents a comprehensive overview of the ocular manifestations of Morquio Syndrome, a rare inherited metabolic disorder distinguished by the deficiency of specific lysosomal enzymes. A thorough examination of the available literature exposes corneal opacification consisting of small gray opacities within the corneal stroma as the most frequently noted ocular finding in Morquio patients. However, these corneal opacities are typically not severe enough to cause significant visual impairment in the majority of patients. Additionally, several other ocular manifestations are noted in the literature, including lens opacities, astigmatism, glaucoma, optic atrophy, corneal thickening, nyctalopia, and hypoesthesia. These findings demonstrate the complexity of ophthalmological involvement in patients with Morquio Syndrome.
ABBREVIATION
| MPS | = Mucopolysaccharidosis |
FUTURE RECOMMENDATIONS
Most of the current studies and reports focused on Morquio A, the more severe and prevalent subtype of Morquio Syndrome. There is a scarcity of existing literature on Morquio B ophthalmological involvement, indicating a need for further research to differentiate and compare the two variants. It is also important to broaden the scope of clinical examinations to look beyond corneal opacification alone as the sole ocular presentation of Morquio Syndrome and assess intraocular pressure, lenticular opacities, nyctalopia, and corneal thickness in more detail. Despite there being no cure for Morquio Syndrome, it is important to investigate new treatment and management approaches to enhance ocular health and quality of life for patients with Morquio Syndrome. The underlying mechanisms responsible for the various ocular manifestations in Morquio Syndrome are also a gap yet to be addressed in the literature. Such advancements in knowledge and intervention would contribute to a more comprehensive approach to the care and management of patients suffering with this rare genetic disorder, enhancing their visual outcomes and overall well-being.
AUTHORS' CONTRIBUTIONS
AR and AFJA were involved in the design, data analysis and interpretation, and drafting of the manuscript. AA, IA, and NS were responsible for editing and revision of the manuscript. All authors read and approved the final manuscript.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

